 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6fty | ||||||
|---|---|---|---|---|---|---|---|
| Title | COMPLEMENT FACTOR D COMPLEXED WITH COMPOUND 5 | ||||||
 Components Components | Complement factor D | ||||||
 Keywords Keywords | HYDROLASE / SERINE PROTEASE | ||||||
| Function / homology |  Function and homology information Function and homology informationcomplement factor D / Alternative complement activation / complement activation, alternative pathway / complement activation / zymogen activation / platelet alpha granule lumen / serine-type peptidase activity / response to bacterium / protein maturation / Platelet degranulation ...complement factor D / Alternative complement activation / complement activation, alternative pathway / complement activation / zymogen activation / platelet alpha granule lumen / serine-type peptidase activity / response to bacterium / protein maturation / Platelet degranulation / secretory granule lumen / ficolin-1-rich granule lumen / serine-type endopeptidase activity / Neutrophil degranulation / proteolysis / : / extracellular exosome / extracellular region Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.67 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.67 Å | ||||||
 Authors Authors | Ostermann, N. | ||||||
 Citation Citation | Journal: ACS Med Chem Lett / Year: 2018 Title: Discovery and Design of First Benzylamine-Based Ligands Binding to an Unlocked Conformation of the Complement Factor D. Authors: Vulpetti, A. / Ostermann, N. / Randl, S. / Yoon, T. / Mac Sweeney, A. / Cumin, F. / Lorthiois, E. / Rudisser, S. / Erbel, P. / Maibaum, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6fty.cif.gz | 63.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6fty.ent.gz | 44 KB | Display | PDB format |
| PDBx/mmJSON format | 6fty.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ft/6ftyftp://data.pdbj.org/pub/pdb/validation_reports/ft/6fty | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6ftzC  6fugC  6fuhC  6fuiC  6fujC  6futC  5fbeS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24739.121 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CFD, DF, PFD / Production host:  |
|---|---|
| #2: Chemical | ChemComp-E7H / 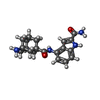  Mass: 352.430 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H24N4O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 352.430 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H24N4O2 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | P2SEQ ANNOTATION (DBREF, SEQADV, MODRES, REMARK 465, COMPND, SOURCE RECORDS) WAS ADDED SEMI- ...P2SEQ ANNOTATION |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.11 Å3/Da / Density % sol: 41.83 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / Details: PEG 3350, 100 mM HEPES pH 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.54 Å |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Sep 3, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.67→53.22 Å / Num. obs: 23224 / % possible obs: 96.2 % / Redundancy: 3.15 % / Biso Wilson estimate: 11.82 Å2 / Rmerge(I) obs: 0.089 / Net I/σ(I): 11.73 |
| Reflection shell | Resolution: 1.67→1.74 Å / Redundancy: 3.02 % / Rmerge(I) obs: 0.189 / Mean I/σ(I) obs: 5.81 / % possible all: 88.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5FBE Resolution: 1.67→53.22 Å / Cor.coef. Fo:Fc: 0.938 / Cor.coef. Fo:Fc free: 0.914 / SU R Cruickshank DPI: 0.103 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.106 / SU Rfree Blow DPI: 0.106 / SU Rfree Cruickshank DPI: 0.105
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.64 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.67→53.22 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.67→1.74 Å / Total num. of bins used: 12
|
