 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6f4r: Human JMJD5 (N308C) in complex with Mn(II), NOG and RCCD1 (139-14... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6f4r | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human JMJD5 (N308C) in complex with Mn(II), NOG and RCCD1 (139-143) (complex-3) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXIDOREDUCTASE / NON-HEME / IRON / 2-OXOGLUTARATE / DIOXYGENASE / JMJC / JMJC DOMAIN / Lysine-specific demethylase 8 / JmjC domain-containing protein 5 / Arginyl C-3 Hydroxylase / JMJD5 / KDM8 / OXYGENASE / HYPOXIA / DNA-BINDING / METAL-BINDING / TRANSLATION / DSBH / FACIAL TRIAD / CYTOPLASM / JMJC HYDROXYLASE / JMJC DEMETHYLASE / KDMS / POST-TRANSLATIONAL MODIFICATIONS / PTM / BETA-HYDROXYLATION / HYDROXYLATION / ARGININE HYDROXYLATION / RCC1 domain-containing protein 1 / RCCD1 / Regulator of chromosome condensation / 40S ribosomal protein S6 / RPS6 / RIBOSOME BIOGENESIS / TRANSCRIPTION / EPIGENETIC REGULATION / SIGNALING / DEVELOPMENT / CELL STRUCTURE / TRANSCRIPTION ACTIVATOR/INHIBITOR / PHOSPHORYLATION / CANCER / POLYMORPHISM | ||||||
| Function / homology |  Function and homology information Function and homology information[protein]-arginine 3-hydroxylase / peptidyl-arginine hydroxylation / peptidyl-arginine 3-dioxygenase activity / histone H3K36 demethylase activity / Hydrolases; Acting on peptide bonds (peptidases) / Protein hydroxylation / aminopeptidase activity / circadian regulation of gene expression / protein destabilization / G2/M transition of mitotic cell cycle ...[protein]-arginine 3-hydroxylase / peptidyl-arginine hydroxylation / peptidyl-arginine 3-dioxygenase activity / histone H3K36 demethylase activity / Hydrolases; Acting on peptide bonds (peptidases) / Protein hydroxylation / aminopeptidase activity / circadian regulation of gene expression / protein destabilization / G2/M transition of mitotic cell cycle / p53 binding / chromosome / chromatin organization / endopeptidase activity / negative regulation of DNA-templated transcription / chromatin binding / positive regulation of DNA-templated transcription / DNA-templated transcription / proteolysis / metal ion binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.3 Å | ||||||
 Authors Authors | Chowdhury, R. / Islam, M.S. / Schofield, C.J. | ||||||
 Citation Citation | Journal: Nat Commun / Year: 2018 Title: JMJD5 is a human arginyl C-3 hydroxylase. Authors: Wilkins, S.E. / Islam, S. / Gannon, J.M. / Markolovic, S. / Hopkinson, R.J. / Ge, W. / Schofield, C.J. / Chowdhury, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6f4r.cif.gz | 191.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6f4r.ent.gz | 153.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6f4r.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f4/6f4rftp://data.pdbj.org/pub/pdb/validation_reports/f4/6f4r | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6f4mC  6f4nC  6f4oC  6f4pSC  6f4qC 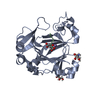 6f4sC  6f4tC C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |

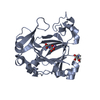








-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein / Protein/peptide , 2 types, 2 molecules AB
| #1: Protein | Mass: 29479.135 Da / Num. of mol.: 1 / Mutation: C217A, C232A, C295A, N308C, C384A Source method: isolated from a genetically manipulated source Details: CATALYTIC DOMAIN (RESIDUES 183-416) / Source: (gene. exp.) Homo sapiens (human) / Gene: KDM8, JMJD5 / Plasmid: PNIC28 BSA4 / Production host:  References: UniProt: Q8N371, [50S ribosomal protein L16]-arginine 3-hydroxylase |
|---|---|
| #2: Protein/peptide | Mass: 583.681 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: FRAGMENT: 139-143 / Source: (synth.) Homo sapiens (human) / References: UniProt: A6NED2 |
-Non-polymers , 4 types, 363 molecules 


| #3: Chemical | ChemComp-MN /  Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn |
|---|---|
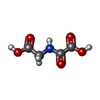
| #4: Chemical | ChemComp-OGA /  Mass: 147.086 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H5NO5 Mass: 147.086 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H5NO5 |
| #5: Chemical | ChemComp-TRS /  Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM |
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 360 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.18 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 5.8 Details: 0.1M Bis-Tris pH 5.8, 25 % PEG3350, 0.002 M MnCl2, 300 nl sitting drops (sample:well, 1:1 ratio) |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9795 Å / Beamline: I03 / Wavelength: 0.9795 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Jan 19, 2016 / Details: MIRRORS |
| Radiation | Collimation: MIRRORS / Monochromator: SI 111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 |
| Reflection | Resolution: 1.3→49.84 Å / Num. obs: 62115 / % possible obs: 100 % / Redundancy: 9 % / Biso Wilson estimate: 15.5 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.079 / Rpim(I) all: 0.031 / Net I/σ(I): 24.3 |
| Reflection shell | Resolution: 1.3→1.35 Å / Redundancy: 8.8 % / Rmerge(I) obs: 1.2 / Mean I/σ(I) obs: 1.7 / Num. unique obs: 6098 / CC1/2: 0.568 / Rpim(I) all: 0.553 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6F4P Resolution: 1.3→49.84 Å / SU ML: 0.12 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 15.5 / Stereochemistry target values: ML Details: REFINEMENT TARGET : ML and BULK SOLVENT MODELLING METHOD USED: FLAT MODEL
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT MODEL / Bsol: 54.2 Å2 / ksol: 0.41 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→49.84 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -39.9383 Å / Origin y: 13.3208 Å / Origin z: 98.6075 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
