 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6f2b: Crystal structure of the complex Fe(II)/alpha-ketoglutarate depen... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6f2b | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the complex Fe(II)/alpha-ketoglutarate dependent dioxygenase KDO1 with Fe(II)/alpha-ketoglutarate | ||||||
 Components Components | L-lysine 3-hydroxylase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Fe(II)/alpha-ketoglutarate / dioxygenases / enzyme / FeII alphaKG form / oxydoreductase | ||||||
| Function / homology | Clavaminate synthase-like / Taurine catabolism dioxygenase TauD, TfdA family / Taurine dioxygenase TauD-like superfamily / 2-oxoglutarate-dependent dioxygenase activity / Oxidoreductases; Acting on paired donors, with incorporation or reduction of molecular oxygen; With 2-oxoglutarate as one donor, and incorporation of one atom of oxygen into each donor / iron ion binding / 2-OXOGLUTARIC ACID / : / L-lysine 3-hydroxylase Function and homology information Function and homology information | ||||||
| Biological species |  Catenulispora acidiphila (bacteria) Catenulispora acidiphila (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Isabet, T. / Stura, E.A. / Legrand, P. / Zaparucha, A. / Bastard, K. | ||||||
 Citation Citation | Journal: Sci Rep / Year: 2018 Title: Structural Studies based on two Lysine Dioxygenases with Distinct Regioselectivity Brings Insights Into Enzyme Specificity within the Clavaminate Synthase-Like Family. Authors: Bastard, K. / Isabet, T. / Stura, E.A. / Legrand, P. / Zaparucha, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6f2b.cif.gz | 536.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6f2b.ent.gz | 441.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6f2b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f2/6f2bftp://data.pdbj.org/pub/pdb/validation_reports/f2/6f2b | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6euoC  6eurC  6exfC  6exhC  6f2aC  6f2eC  6f6jC  6f9pC  2wbqS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39236.211 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Catenulispora acidiphila (strain DSM 44928 / NRRL B-24433 / NBRC 102108 / JCM 14897) (bacteria)Gene: Caci_0231 / Production host: References: UniProt: C7QJ42, Oxidoreductases; Acting on paired donors, with incorporation or reduction of molecular oxygen; With 2-oxoglutarate as one donor, and incorporation of one atom of oxygen into each donor #2: Chemical | ChemComp-FE2 /   Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe#3: Chemical | ChemComp-AKG / 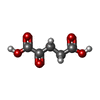  Mass: 146.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C5H6O5 Mass: 146.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C5H6O5#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1163 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1163 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.79 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: KDO1: 9.3 mg/ml in 0.2 M NaCl, .001 M DTT, 0.05 M Tris-HCl, pH 8 Precipitant: 18% PEG3350, 0.15 M Tris pH 7.5, 0.3 M Na Acetate. Soaking/cryo : 20% PEG3350, 0.15 M Tris pH 7.5, 0.2 M NaCl, 0. ...Details: KDO1: 9.3 mg/ml in 0.2 M NaCl, .001 M DTT, 0.05 M Tris-HCl, pH 8 Precipitant: 18% PEG3350, 0.15 M Tris pH 7.5, 0.3 M Na Acetate. Soaking/cryo : 20% PEG3350, 0.15 M Tris pH 7.5, 0.2 M NaCl, 0.02 M Na Succinate, 0.001 M FeIISO4 (0.5 M stock solution prepared in 0.050 M dithionite), 0.05 M alpha-ketoglutarate, 20% glycerol, 10 minute soaking PH range: 7.5-8.0 / Temp details: air conditioned room |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 1 / Wavelength: 0.97857 Å / Beamline: PROXIMA 1 / Wavelength: 0.97857 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Mar 26, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. obs: 101974 / % possible obs: 98.8 % / Redundancy: 3.4 % / Biso Wilson estimate: 38.04 Å2 / Rrim(I) all: 0.01 / Net I/σ(I): 10.86 |
| Reflection shell | Resolution: 2→2.05 Å / Redundancy: 3.4 % / Num. unique obs: 7151 / Rrim(I) all: 0.1125 / % possible all: 93.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2WBQ Resolution: 2→40 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.941 / SU R Cruickshank DPI: 0.153 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.159 / SU Rfree Blow DPI: 0.136 / SU Rfree Cruickshank DPI: 0.134
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 45.56 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.23 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2→40 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.05 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
