 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6eom | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of DPP III from Caldithrix abyssi | ||||||
 Components Components | MutT/NUDIX family protein | ||||||
 Keywords Keywords | HYDROLASE / Caldithrix abyssi / Metallopeptidase / Dipeptidyl peptidase III / DPP III / Zinc-Hydrolase | ||||||
| Function / homology | Peptidase family M49 / Peptidase family M49 / dipeptidyl-peptidase activity / Prokaryotic membrane lipoprotein lipid attachment site profile. / metal ion binding / cytoplasm / ALANINE / LYSINE / MutT/NUDIX family protein Function and homology information Function and homology information | ||||||
| Biological species |  Caldithrix abyssi DSM 13497 (bacteria) Caldithrix abyssi DSM 13497 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.103 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.103 Å | ||||||
 Authors Authors | Sabljic, I. | ||||||
| Funding support |  Croatia, 1items Croatia, 1items
| ||||||
 Citation Citation | Journal: PLoS ONE / Year: 2018 Title: The first dipeptidyl peptidase III from a thermophile: Structural basis for thermal stability and reduced activity. Authors: Sabljic, I. / Tomin, M. / Matovina, M. / Sucec, I. / Tomasic Paic, A. / Tomic, A. / Abramic, M. / Tomic, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6eom.cif.gz | 233.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6eom.ent.gz | 184.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6eom.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eo/6eomftp://data.pdbj.org/pub/pdb/validation_reports/eo/6eom | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 65511.031 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Caldithrix abyssi DSM 13497 (bacteria) / Gene: Cabys_2252, Calab_3349 / Plasmid: pET-21a(+) / Production host: |
|---|
-Non-polymers , 7 types, 342 molecules 


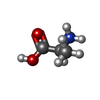


| #2: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #3: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl | ||||||||
| #4: Chemical |  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#5: Chemical | ChemComp-ALA / |  Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2#6: Chemical | ChemComp-LYS / |  Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N2O2 Type: L-peptide linking / Mass: 147.195 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15N2O2#7: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 335 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.37 Å3/Da / Density % sol: 48.02 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 6.5 / Details: 0.1 M Bis-Tris pH 6.5, 25% PEG 3,350 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 1.2705 Å / Beamline: 5.2R / Wavelength: 1.2705 Å | ||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 2M / Detector: PIXEL / Date: Nov 3, 2014 | ||||||||||||||||||||||||
| Radiation | Monochromator: Double Crystal Si111 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.2705 Å / Relative weight: 1 | ||||||||||||||||||||||||
| Reflection | Resolution: 2.1→48 Å / Num. obs: 36965 / % possible obs: 99.8 % / Redundancy: 8.7 % / Biso Wilson estimate: 29.44 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.117 / Rpim(I) all: 0.042 / Rrim(I) all: 0.124 / Net I/σ(I): 13.4 | ||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Se-Met labeled protein solved using MAD metode Resolution: 2.103→48 Å / SU ML: 0.28 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 29.42
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 89.93 Å2 / Biso mean: 38.3051 Å2 / Biso min: 19.87 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.103→48 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 13
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
