histone H2AX kinase activity / Golgi disassembly / Cajal body organization / histone H3T3 kinase activity / Nuclear Envelope Breakdown / positive regulation of protein localization to chromatin / mitotic nuclear membrane disassembly / histone H3S10 kinase activity / regulation of neuron migration / Golgi stack ...histone H2AX kinase activity / Golgi disassembly / Cajal body organization / histone H3T3 kinase activity / Nuclear Envelope Breakdown / positive regulation of protein localization to chromatin / mitotic nuclear membrane disassembly / histone H3S10 kinase activity / regulation of neuron migration / Golgi stack / Initiation of Nuclear Envelope (NE) Reformation / nucleosomal DNA binding / Cajal body / neuron projection development / kinase activity / protein autophosphorylation / histone binding / protein phosphorylation / protein kinase activity / non-specific serine/threonine protein kinase / chromatin remodeling / protein serine kinase activity / cell division / protein serine/threonine kinase activity / DNA damage response / protein kinase binding / chromatin / nucleolus / signal transduction / nucleoplasm / ATP binding / nucleus / cytoplasm / cytosol Similarity search - Function
: / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. ...: / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / Orthogonal Bundle / Mainly Alpha Similarity search - Domain/homology
Resolution: 2.1→33.96 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.942 / SU B: 11.567 / SU ML: 0.145 / Cross valid method: THROUGHOUT / ESU R: 0.183 / ESU R Free: 0.159 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.22555
5033
5.1 %
RANDOM
Rwork
0.19463
-
-
-
obs
0.19622
94402
99.92 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Brazil, 1items
Brazil, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj

 Assembly
Assembly










 Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl Mass: 59.044 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H3O2
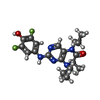
Mass: 59.044 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 391.415 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H23F2N5O2
Mass: 391.415 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H23F2N5O2 Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: SO4 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Sample preparation
Sample preparation / Beamline: I24 / Wavelength: 0.96861 Å
/ Beamline: I24 / Wavelength: 0.96861 Å Processing
Processing