 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6cee: Crystal structure of fragment 3-(1-Methyl-2-oxo-1,2-dihydroquinox... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6cee | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of fragment 3-(1-Methyl-2-oxo-1,2-dihydroquinoxalin-3-yl)propionic acid bound in the ubiquitin binding pocket of the HDAC6 zinc-finger domain | ||||||
 Components Components | Histone deacetylase 6 | ||||||
 Keywords Keywords | HYDROLASE / HISTONE DEACETYLASE / HDAC / HDAC6 / UBIQUITIN / STRUCTURAL GENOMICS CONSORTIUM / SGC | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of hydrogen peroxide metabolic process / cellular response to topologically incorrect protein / polyubiquitinated misfolded protein transport / negative regulation of aggrephagy / response to misfolded protein / positive regulation of protein oligomerization / type 2 mitophagy / negative regulation of protein-containing complex disassembly / peroxidase inhibitor activity / regulation of autophagy of mitochondrion ...negative regulation of hydrogen peroxide metabolic process / cellular response to topologically incorrect protein / polyubiquitinated misfolded protein transport / negative regulation of aggrephagy / response to misfolded protein / positive regulation of protein oligomerization / type 2 mitophagy / negative regulation of protein-containing complex disassembly / peroxidase inhibitor activity / regulation of autophagy of mitochondrion / erythrocyte enucleation / protein-containing complex disassembly / tubulin deacetylation / Cilium Assembly / collateral sprouting / regulation of establishment of protein localization / deacetylase activity / Transcriptional regulation by RUNX2 / tubulin deacetylase activity / lysosome localization / negative regulation of microtubule depolymerization / cilium disassembly / ATPase inhibitor activity / protein deacetylation / misfolded protein binding / positive regulation of type 2 mitophagy / dendritic spine morphogenesis / ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway / cytoplasmic ubiquitin ligase complex / regulation of androgen receptor signaling pathway / aggresome assembly / Transferases; Acyltransferases; Aminoacyltransferases / regulation of fat cell differentiation / histone deacetylase activity / cellular response to misfolded protein / protein lysine deacetylase activity / aggresome / Hydrolases; Acting on carbon-nitrogen bonds, other than peptide bonds; In linear amides / microtubule associated complex / positive regulation of intracellular estrogen receptor signaling pathway / Notch-HLH transcription pathway / axonal transport of mitochondrion / RUNX2 regulates osteoblast differentiation / regulation of microtubule-based movement / negative regulation of gene expression, epigenetic / histone deacetylase complex / cell leading edge / protein quality control for misfolded or incompletely synthesized proteins / dynein complex binding / positive regulation of epithelial cell migration / cilium assembly / regulation of macroautophagy / HSF1 activation / polyubiquitin modification-dependent protein binding / alpha-tubulin binding / negative regulation of protein-containing complex assembly / beta-tubulin binding / negative regulation of proteolysis / multivesicular body / axon cytoplasm / inclusion body / transcription corepressor binding / regulation of autophagy / actin filament organization / ubiquitin binding / regulation of protein stability / Hsp90 protein binding / intracellular protein transport / Late endosomal microautophagy / caveola / beta-catenin binding / protein destabilization / NOTCH1 Intracellular Domain Regulates Transcription / tau protein binding / Constitutive Signaling by NOTCH1 PEST Domain Mutants / Constitutive Signaling by NOTCH1 HD+PEST Domain Mutants / epidermal growth factor receptor signaling pathway / histone deacetylase binding / cellular response to hydrogen peroxide / Chaperone Mediated Autophagy / protein polyubiquitination / Aggrephagy / ubiquitin protein ligase activity / cellular response to heat / actin binding / microtubule binding / microtubule / perikaryon / ciliary basal body / RNA polymerase II cis-regulatory region sequence-specific DNA binding / axon / negative regulation of DNA-templated transcription / ubiquitin protein ligase binding / centrosome / dendrite / perinuclear region of cytoplasm / enzyme binding / zinc ion binding / nucleoplasm / nucleus Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.55 Å X-RAY DIFFRACTION / Resolution: 1.55 Å | ||||||
 Authors Authors | Harding, R.J. / Halabelian, L. / Ferreira de Freitas, R. / Franzoni, I. / Ravichandran, M. / Lautens, M. / Santhakumar, V. / Schapira, M. / Bountra, C. / Edwards, A.M. ...Harding, R.J. / Halabelian, L. / Ferreira de Freitas, R. / Franzoni, I. / Ravichandran, M. / Lautens, M. / Santhakumar, V. / Schapira, M. / Bountra, C. / Edwards, A.M. / Arrowsmith, C.M. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: J. Med. Chem. / Year: 2018 Title: Identification and Structure-Activity Relationship of HDAC6 Zinc-Finger Ubiquitin Binding Domain Inhibitors. Authors: Ferreira de Freitas, R. / Harding, R.J. / Franzoni, I. / Ravichandran, M. / Mann, M.K. / Ouyang, H. / Lautens, M. / Santhakumar, V. / Arrowsmith, C.H. / Schapira, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6cee.cif.gz | 38.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6cee.ent.gz | 23.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6cee.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ce/6ceeftp://data.pdbj.org/pub/pdb/validation_reports/ce/6cee | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6ce6C  6ce8C  6ceaC  6cecC  6cedC  6cefC  5kh3S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 11932.607 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HDAC6, KIAA0901, JM21 / Plasmid: pET28-lic / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-EYM / | 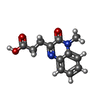  Mass: 232.235 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H12N2O3 Mass: 232.235 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H12N2O3#4: Chemical | ChemComp-UNX /   Num. of mol.: 4 / Source method: obtained synthetically Num. of mol.: 4 / Source method: obtained synthetically#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 87 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 87 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.09 Å3/Da / Density % sol: 41.07 % / Mosaicity: 0 ° |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 4.6 Details: 2 M Na-formate, 0.2 M Na-acetate pH4.6, 5 % ethylene glycol |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E SUPERBRIGHT / Wavelength: 1.54178 Å | ||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU SATURN A200 / Detector: CCD / Date: Aug 24, 2017 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.55→30 Å / Num. obs: 15166 / % possible obs: 100 % / Redundancy: 6.7 % / CC1/2: 1 / Rmerge(I) obs: 0.037 / Rpim(I) all: 0.015 / Rrim(I) all: 0.04 / Net I/σ(I): 40.5 / Num. measured all: 101184 / Scaling rejects: 3 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: pdbid 5KH3 Resolution: 1.55→30 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.959 / SU B: 1.024 / SU ML: 0.038 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.069 / ESU R Free: 0.067 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: Users of this crystal structure: verify our intepretion of the electron density. Amplitudes and unmerged intensities are included with this deposition. Diffraction images will be deposited ...Details: Users of this crystal structure: verify our intepretion of the electron density. Amplitudes and unmerged intensities are included with this deposition. Diffraction images will be deposited at a later date in a public repository. Geometry restraints for the ligand were prepared with GRADE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 36.3 Å2 / Biso mean: 12.359 Å2 / Biso min: 6.93 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.55→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.55→1.59 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
