regulation of protein kinase activity / positive regulation of autophagy of mitochondrion / Ca2+/calmodulin-dependent protein kinase / CAMKK-AMPK signaling cascade / calcium/calmodulin-dependent protein kinase activity / CREB1 phosphorylation through the activation of CaMKII/CaMKK/CaMKIV cascasde / CaMK IV-mediated phosphorylation of CREB / Activation of RAC1 downstream of NMDARs / Activation of AMPK downstream of NMDARs / cellular response to reactive oxygen species ...regulation of protein kinase activity / positive regulation of autophagy of mitochondrion / Ca2+/calmodulin-dependent protein kinase / CAMKK-AMPK signaling cascade / calcium/calmodulin-dependent protein kinase activity / CREB1 phosphorylation through the activation of CaMKII/CaMKK/CaMKIV cascasde / CaMK IV-mediated phosphorylation of CREB / Activation of RAC1 downstream of NMDARs / Activation of AMPK downstream of NMDARs / cellular response to reactive oxygen species / calcium-mediated signaling / protein autophosphorylation / MAPK cascade / protein tyrosine kinase activity / protein phosphorylation / calmodulin binding / neuron projection / protein serine kinase activity / protein serine/threonine kinase activity / calcium ion binding / positive regulation of DNA-templated transcription / nucleoplasm / ATP binding / cytosol Similarity search - Function
Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily Similarity search - Domain/homology
Resolution: 1.8→19.88 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.941 / SU B: 4.882 / SU ML: 0.08 / Cross valid method: THROUGHOUT / ESU R: 0.119 / ESU R Free: 0.118 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.22768
1539
5 %
RANDOM
Rwork
0.19064
-
-
-
obs
0.19245
29482
99.88 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Brazil, 1items
Brazil, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj




 Assembly
Assembly

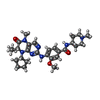
 Mass: 521.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H39N7O3
Mass: 521.654 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H39N7O3
 Mass: 59.044 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H3O2
Mass: 59.044 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 24-ID-C / Wavelength: 0.9791 Å
/ Beamline: 24-ID-C / Wavelength: 0.9791 Å Processing
Processing