 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6bg3: Structure of (3S,4S)-1-benzyl-4-(3-(3-(trifluoromethyl)phenyl)ure... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6bg3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of (3S,4S)-1-benzyl-4-(3-(3-(trifluoromethyl)phenyl)ureido)piperidin-3-yl acetate bound to DCN1 | ||||||
 Components Components | Endolysin, DCN1-like protein 1 chimera | ||||||
 Keywords Keywords | HYDROLASE / LIGASE/INHIBITOR / E3 Ligase / LIGASE-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of protein neddylation / ubiquitin-like protein binding / regulation of protein neddylation / protein neddylation / ubiquitin conjugating enzyme binding / cullin family protein binding / regulation of protein ubiquitination / ubiquitin ligase complex / viral release from host cell by cytolysis / peptidoglycan catabolic process ...positive regulation of protein neddylation / ubiquitin-like protein binding / regulation of protein neddylation / protein neddylation / ubiquitin conjugating enzyme binding / cullin family protein binding / regulation of protein ubiquitination / ubiquitin ligase complex / viral release from host cell by cytolysis / peptidoglycan catabolic process / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / Neddylation / host cell cytoplasm / defense response to bacterium / nucleoplasm / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.05 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.05 Å | ||||||
 Authors Authors | Guy, R.K. / Schulman, B.A. / Scott, D.C. / Hammill, J.T. | ||||||
 Citation Citation | Journal: J. Med. Chem. / Year: 2018 Title: Piperidinyl Ureas Chemically Control Defective in Cullin Neddylation 1 (DCN1)-Mediated Cullin Neddylation. Authors: Hammill, J.T. / Scott, D.C. / Min, J. / Connelly, M.C. / Holbrook, G. / Zhu, F. / Matheny, A. / Yang, L. / Singh, B. / Schulman, B.A. / Guy, R.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6bg3.cif.gz | 299.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6bg3.ent.gz | 233.1 KB | Display | PDB format |
| PDBx/mmJSON format | 6bg3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6bg3_validation.pdf.gz | 736.5 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6bg3_full_validation.pdf.gz | 738.7 KB | Display | |
| Data in XML | 6bg3_validation.xml.gz | 22.5 KB | Display | |
| Data in CIF | 6bg3_validation.cif.gz | 36.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bg/6bg3ftp://data.pdbj.org/pub/pdb/validation_reports/bg/6bg3 | HTTPS FTP |
-Related structure data
| Related structure data |  6bg5C  2lzmS  3tduS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 44307.352 Da / Num. of mol.: 1 / Fragment: PONY Source method: isolated from a genetically manipulated source Source: (gene. exp.) Enterobacteria phage T4 (virus), (gene. exp.) Homo sapiens (human)Gene: E, DCUN1D1, DCUN1L1, RP42, SCCRO / Plasmid: pRSF DUET / Production host:  |
|---|---|
| #2: Chemical | ChemComp-DOJ / 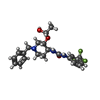  Mass: 437.455 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H26F3N3O3 Mass: 437.455 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H26F3N3O3 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 678 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 678 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.53 % / Mosaicity: 0.22 ° |
|---|---|
| Crystal grow | Temperature: 277 K / Method: evaporation / Details: 6% PEG3350, 0.2M NH4Br |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 0.9795 Å / Beamline: 24-ID-C / Wavelength: 0.9795 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Apr 12, 2014 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9795 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.05→50 Å / Num. obs: 176138 / % possible obs: 98.9 % / Redundancy: 3.2 % / Rmerge(I) obs: 0.032 / Rpim(I) all: 0.021 / Rrim(I) all: 0.039 / Χ2: 0.937 / Net I/σ(I): 10.2 / Num. measured all: 571480 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2LZM, 3TDU Resolution: 1.05→49.279 Å / SU ML: 0.11 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 16.75
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 66.86 Å2 / Biso mean: 21.7765 Å2 / Biso min: 8.53 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.05→49.279 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 30
|
