| Entry | Database: PDB / ID: 5zas
|
|---|
| Title | Crystal structure of 5-formylcytosine containing decamer dsDNA |
|---|
 Components Components | DNA (5'-D(*CP*CP*AP*GP*(5FC)P*GP*CP*TP*GP*G)-3')  Keywords Keywords | DNA / Cytosine modification |
|---|
| Function / homology | BICARBONATE ION / DNA Function and homology information Function and homology information |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.56 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.56 Å |
|---|
 Authors Authors | Fu, T.R. / Zhang, L. |
|---|
 Citation Citation | Journal: Chem Sci / Year: 2019
Title: Thymine DNA glycosylase recognizes the geometry alteration of minor grooves induced by 5-formylcytosine and 5-carboxylcytosine.
Authors: Fu, T. / Liu, L. / Yang, Q.L. / Wang, Y. / Xu, P. / Zhang, L. / Liu, S. / Dai, Q. / Ji, Q. / Xu, G.L. / He, C. / Luo, C. / Zhang, L. |
|---|
| History | | Deposition | Feb 8, 2018 | Deposition site: PDBJ / Processing site: PDBJ |
|---|
| Revision 1.0 | Feb 13, 2019 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Aug 28, 2019 | Group: Data collection / Database references / Category: citation / citation_author
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.title / _citation.year |
|---|
| Revision 1.2 | Sep 18, 2019 | Group: Data collection / Database references / Category: citation / citation_author
Item: _citation.journal_id_ISSN / _citation.journal_volume ..._citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_PubMed / _citation.title / _citation_author.identifier_ORCID / _citation_author.name |
|---|
| Revision 1.3 | Mar 27, 2024 | Group: Data collection / Database references / Category: chem_comp_atom / chem_comp_bond / database_2
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj




 Assembly
Assembly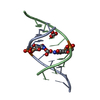
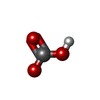
 Mass: 61.017 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM
Mass: 61.017 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CHO3 / Comment: pH buffer*YM Mass: 18.015 Da / Num. of mol.: 118 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 118 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL19U1 / Wavelength: 0.9676 Å
/ Beamline: BL19U1 / Wavelength: 0.9676 Å Processing
Processing