Entry Database : PDB / ID : 5yo1Title Structure of ePepN E298A mutant in complex with Puromycin Aminopeptidase N Keywords / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Escherichia coli K12 (bacteria)Method / / Resolution : 2.5 Å Authors Ganji, R.J. / Reddi, R. / Marapaka, A.K. / Addlagatta, A. Funding support Organization Grant number Country BT-BRB-TF-2-2011
Journal : Int.J.Biol.Macromol. / Year : 2020Title : Puromycin, a selective inhibitor of PSA acts as a substrate for other M1 family aminopeptidases: Biochemical and structural basisAuthors : Reddi, R. / Ganji, R.J. / Marapaka, A.K. / Bala, S.C. / Yerra, N.V. / Haque, N. / Addlagatta, A. History Deposition Oct 26, 2017 Deposition site / Processing site Revision 1.0 Nov 7, 2018 Provider / Type Revision 1.1 Jul 27, 2022 Group / Category / citation_author / database_2Item _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_ASTM / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.title / _citation.year / _database_2.pdbx_DOI / _database_2.pdbx_database_accession Revision 1.2 Nov 22, 2023 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_model
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors India, 1items
India, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj Assembly
Assembly




 Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn
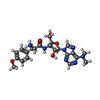
Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Type: RNA linking / Mass: 471.510 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H29N7O5 / Feature type: SUBJECT OF INVESTIGATION
Type: RNA linking / Mass: 471.510 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H29N7O5 / Feature type: SUBJECT OF INVESTIGATION Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 92.094 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: C3H8O3 Sample preparation
Sample preparation Processing
Processing