| Entry | Database: PDB / ID: 5xz4
|
|---|
| Title | The X-tay structure of Bumblebee PGRP-SA |
|---|
 Components Components | Bumblebee peptidoglycan recognition protein SA |
|---|
 Keywords Keywords | IMMUNE SYSTEM / Bumblebee / PGRP-SA |
|---|
| Function / homology |  Function and homology information Function and homology information
N-acetylmuramoyl-L-alanine amidase / N-acetylmuramoyl-L-alanine amidase activity / peptidoglycan binding / peptidoglycan catabolic process / innate immune response / zinc ion bindingSimilarity search - Function Peptidoglycan recognition protein, PGRP-S / Peptidoglycan recognition protein family domain, metazoa/bacteria / Peptidoglycan recognition protein / Animal peptidoglycan recognition proteins homologous to Bacteriophage T3 lysozyme. / Lysozyme-like / Peptidoglycan recognition protein-like / Ami_2 / N-acetylmuramoyl-L-alanine amidase / N-acetylmuramoyl-L-alanine amidase domain / N-acetylmuramoyl-L-alanine amidase/PGRP domain superfamily ...Peptidoglycan recognition protein, PGRP-S / Peptidoglycan recognition protein family domain, metazoa/bacteria / Peptidoglycan recognition protein / Animal peptidoglycan recognition proteins homologous to Bacteriophage T3 lysozyme. / Lysozyme-like / Peptidoglycan recognition protein-like / Ami_2 / N-acetylmuramoyl-L-alanine amidase / N-acetylmuramoyl-L-alanine amidase domain / N-acetylmuramoyl-L-alanine amidase/PGRP domain superfamily / 3-Layer(aba) Sandwich / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Bombus (bumble bees) Bombus (bumble bees) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.41 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.41 Å |
|---|
 Authors Authors | Liu, Y.J. / Huang, J.X. / Zhao, X.M. / An, J.D. |
|---|
| Funding support |  China, 2items China, 2items | Organization | Grant number | Country |
|---|
| Natural Science Foundation of China | NO.31672500 | China | | Agricultural Science and Technology Innovation Program | CAAS-ASTIP-2015-IAR | China |
|
|---|
 Citation Citation | Journal: J Immunol. / Year: 2019
Title: Structural Insights into the Preferential Binding of PGRP-SAs from Bumblebees and Honeybees to Dap-Type Peptidoglycans Rather than Lys-Type Peptidoglycans.
Authors: Liu, Y.J. / Zhao, X.M. / Huang, J.X. / Chen, M.M. / An, J.D. |
|---|
| History | | Deposition | Jul 11, 2017 | Deposition site: PDBJ / Processing site: PDBJ |
|---|
| Revision 1.0 | Sep 19, 2018 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Oct 2, 2019 | Group: Data collection / Database references / Category: citation / citation_author
Item: _citation.country / _citation.journal_abbrev ..._citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year |
|---|
| Revision 1.2 | Nov 22, 2023 | Group: Data collection / Database references / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads




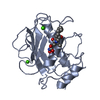




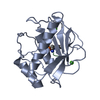
 PDBj
PDBj Assembly
Assembly
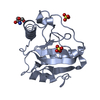



 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4
 Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM
Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 18.015 Da / Num. of mol.: 338 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 338 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing