 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5ts8: Z. MAYS CK2 KINASE ALPHA SUBUNIT IN COMPLEX WITH THE ATP-COMPETIT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5ts8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Z. MAYS CK2 KINASE ALPHA SUBUNIT IN COMPLEX WITH THE ATP-COMPETITIVE INHIBITOR 5,6-DIBROMOBENZOTRIAZOLE | |||||||||
 Components Components | Protein kinase CK2 catalytic subunit CK2 alpha-3 | |||||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / CK2 / CASEIN KINASE 2 / INHIBITOR / BROMO-BENZOTRIAZOLE / TRANSFERASE-TRANSFERASE INHIBITOR COMPLEX / HALOGEN BOND | |||||||||
| Function / homology |  Function and homology information Function and homology informationphotoperiodism / inflorescence development / protein kinase CK2 complex / non-specific serine/threonine protein kinase / regulation of cell cycle / protein serine kinase activity / protein serine/threonine kinase activity / DNA damage response / ATP binding / nucleus / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | |||||||||
 Authors Authors | Winiewska, M. / Kucinska, K. / Czapinska, H. / Piasecka, A. / Bochtler, M. / Poznanski, J. | |||||||||
| Funding support |  Poland, 2items Poland, 2items
| |||||||||
 Citation Citation | Journal: To Be Published Title: STRUCTURAL AND THERMODYNAMIC ANALYSIS OF 5,6-DIBROMOBENZOTRIAZOLE BINDING TO CASEIN KINASE 2 ALPHA Authors: Winiewska, M. / Kucinska, K. / Czapinska, H. / Piasecka, A. / Bochtler, M. / Poznanski, J. #1: Journal: Biochim. Biophys. Acta / Year: 2015 Title: Thermodynamics parameters for binding of halogenated benzotriazole inhibitors of human protein kinase CK2alpha. Authors: Winiewska, M. / Kucinska, K. / Makowska, M. / Poznanski, J. / Shugar, D. #2: Journal: Biochem. Biophys. Res. Commun. / Year: 2015 Title: Thermodynamic parameters for binding of some halogenated inhibitors of human protein kinase CK2. Authors: Winiewska, M. / Makowska, M. / Maj, P. / Wielechowska, M. / Bretner, M. / Poznanski, J. / Shugar, D. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5ts8.cif.gz | 182.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5ts8.ent.gz | 144.1 KB | Display | PDB format |
| PDBx/mmJSON format | 5ts8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ts/5ts8ftp://data.pdbj.org/pub/pdb/validation_reports/ts/5ts8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4rlkS S: Starting model for refinement |
|---|---|
| Similar structure data |
















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 40436.230 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|
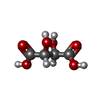
-Non-polymers , 9 types, 463 molecules 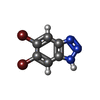







| #2: Chemical | ChemComp-7M0 /  Mass: 276.916 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H3Br2N3 Mass: 276.916 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H3Br2N3 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #3: Chemical |  Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6#4: Chemical | ChemComp-FMT / |  Mass: 46.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH2O2 Mass: 46.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH2O2#5: Chemical | ChemComp-DMS / |  Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#6: Chemical | ChemComp-ACT / |  Mass: 59.044 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C2H3O2#7: Chemical |  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#8: Chemical | ChemComp-BME / |  Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS#9: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 453 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Compound details | THE ACTIVE FORM OF THE PROTEIN KINASE CK2 IS HETEROTETRAMERIC AND COMPOSED OF TWO ALPHA SUBUNITS ...THE ACTIVE FORM OF THE PROTEIN KINASE CK2 IS HETEROTETR |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.82 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: pH 7.5 (1M Sodium HEPES; MOPS), 40% v/v PEG 500* MME; 20 % w/v PEG 20K; 0.2M Sodium formate; 0.2M Ammonium acetate; 0.2M Sodium citrate tribasic dihydrate; 0.2M Sodium potassium tartrate ...Details: pH 7.5 (1M Sodium HEPES; MOPS), 40% v/v PEG 500* MME; 20 % w/v PEG 20K; 0.2M Sodium formate; 0.2M Ammonium acetate; 0.2M Sodium citrate tribasic dihydrate; 0.2M Sodium potassium tartrate tetrahydrate; 0.2M Sodium oxamate PH range: 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY  / Beamline: P14 (MX2) / Wavelength: 0.9116 Å / Beamline: P14 (MX2) / Wavelength: 0.9116 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Aug 6, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9116 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→51.96 Å / Num. obs: 60461 / % possible obs: 98 % / Redundancy: 3.23 % / Biso Wilson estimate: 22.5 Å2 / CC1/2: 0.99 / Rmerge(I) obs: 0.057 / Rsym value: 0.048 / Net I/σ(I): 14.19 |
| Reflection shell | Resolution: 1.45→1.54 Å / Redundancy: 2.78 % / Rmerge(I) obs: 0.55 / Mean I/σ(I) obs: 2.81 / CC1/2: 0.84 / % possible all: 95.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4RLK Resolution: 1.45→51.96 Å / Cor.coef. Fo:Fc: 0.976 / Cor.coef. Fo:Fc free: 0.971 / SU B: 2.551 / SU ML: 0.047 / Cross valid method: THROUGHOUT / ESU R: 0.066 / ESU R Free: 0.065 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. TLS HAS BEEN USED AND U VALUES GENERATED WITH TLS CONTRIBUTION ADDED. DIFFERENCE DENSITY NEXT TO THE OH OXYGEN ATOM OF TYR257 AND TYR26 ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. TLS HAS BEEN USED AND U VALUES GENERATED WITH TLS CONTRIBUTION ADDED. DIFFERENCE DENSITY NEXT TO THE OH OXYGEN ATOM OF TYR257 AND TYR26 MIGHT INDICATE THE X-RAY RADIATION INDUCED HYDROPEROXIDE FORMATION. THE COMPLICATED COMPOSITION OF THE CRYSTALLIZATION BUFFER PRECLUDED UNAMBIGUOUS IDENTIFICATION OF THE LIGANDS. IN SOME CASES MIXTURES OF DIFFERENT COMPOUNDS ARE POSSIBLE EG TARTRATE, OXAMATE, ACETATE (AND DMSO), IN OTHER CASES THE LIGAND IDENTITY COULD NOT BE GUESSED AND THE DIFFERENCE DENSITY COULD AT BEST BE MODELLED AS A SET OF DISORDERED WATER MOLECULES.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.744 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→51.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|