- PDB-5ohq: Crystal structure of the KOW6-KOW7 domain of human DSIF -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 5ohq
Title
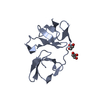
Crystal structure of the KOW6-KOW7 domain of human DSIF
Components
Transcription elongation factor SPT5
Keywords
TRANSCRIPTION / RNA polymerase II / transcription elongation
Function / homology
Function and homology information
negative regulation of DNA-templated transcription, elongation / DSIF complex / regulation of transcription elongation by RNA polymerase II / positive regulation of DNA-templated transcription, elongation / Abortive elongation of HIV-1 transcript in the absence of Tat / RNA Pol II CTD phosphorylation and interaction with CE during HIV infection / RNA Pol II CTD phosphorylation and interaction with CE / Formation of the Early Elongation Complex / Formation of the HIV-1 Early Elongation Complex / mRNA Capping ...negative regulation of DNA-templated transcription, elongation / DSIF complex / regulation of transcription elongation by RNA polymerase II / positive regulation of DNA-templated transcription, elongation / Abortive elongation of HIV-1 transcript in the absence of Tat / RNA Pol II CTD phosphorylation and interaction with CE during HIV infection / RNA Pol II CTD phosphorylation and interaction with CE / Formation of the Early Elongation Complex / Formation of the HIV-1 Early Elongation Complex / mRNA Capping / positive regulation of macroautophagy / RNA polymerase II transcribes snRNA genes / Pausing and recovery of Tat-mediated HIV elongation / Tat-mediated HIV elongation arrest and recovery / HIV elongation arrest and recovery / Pausing and recovery of HIV elongation / Tat-mediated elongation of the HIV-1 transcript / Formation of HIV-1 elongation complex containing HIV-1 Tat / Formation of HIV elongation complex in the absence of HIV Tat / RNA Polymerase II Transcription Elongation / Formation of RNA Pol II elongation complex / RNA Polymerase II Pre-transcription Events / TP53 Regulates Transcription of DNA Repair Genes / positive regulation of transcription elongation by RNA polymerase II / transcription elongation by RNA polymerase II / protein heterodimerization activity / mRNA binding / chromatin binding / negative regulation of transcription by RNA polymerase II / enzyme binding / positive regulation of transcription by RNA polymerase II / RNA binding / nucleoplasm / nucleus Similarity search - Function
Journal: Nat Struct Mol Biol / Year: 2017 Title: Structure of a transcribing RNA polymerase II-DSIF complex reveals a multidentate DNA-RNA clamp. Authors: Carrie Bernecky / Jürgen M Plitzko / Patrick Cramer / Abstract: During transcription, RNA polymerase II (Pol II) associates with the conserved elongation factor DSIF. DSIF renders the elongation complex stable and functions during Pol II pausing and RNA ...During transcription, RNA polymerase II (Pol II) associates with the conserved elongation factor DSIF. DSIF renders the elongation complex stable and functions during Pol II pausing and RNA processing. We combined cryo-EM and X-ray crystallography to determine the structure of the mammalian Pol II-DSIF elongation complex at a nominal resolution of 3.4 Å. Human DSIF has a modular structure with two domains forming a DNA clamp, two domains forming an RNA clamp, and one domain buttressing the RNA clamp. The clamps maintain the transcription bubble, position upstream DNA, and retain the RNA transcript in the exit tunnel. The mobile C-terminal region of DSIF is located near exiting RNA, where it can recruit factors for RNA processing. The structure provides insight into the roles of DSIF during mRNA synthesis.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 3items
Germany, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads















 PDBj
PDBj









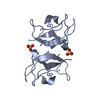
 Assembly
Assembly



 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
 Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 18.015 Da / Num. of mol.: 238 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 238 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X06SA / Wavelength: 0.999987357022 Å
/ Beamline: X06SA / Wavelength: 0.999987357022 Å Processing
Processing