 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5n32: cAMP-dependent Protein Kinase A from Cricetulus griseus in comple... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5n32 | ||||||
|---|---|---|---|---|---|---|---|
| Title | cAMP-dependent Protein Kinase A from Cricetulus griseus in complex with fragment like molecule 4-chlorobenzyl carbamimidothioate | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE / fragment / complex / serine threonine kinase / cAMP / kinase / PKA | ||||||
| Function / homology |  Function and homology information Function and homology informationcAMP-dependent protein kinase inhibitor activity / histone H1-4S35 kinase activity / cAMP-dependent protein kinase / regulation of protein processing / dorsal/ventral neural tube patterning / cAMP-dependent protein kinase activity / cellular response to parathyroid hormone stimulus / protein localization to lipid droplet / regulation of bicellular tight junction assembly / cAMP-dependent protein kinase complex ...cAMP-dependent protein kinase inhibitor activity / histone H1-4S35 kinase activity / cAMP-dependent protein kinase / regulation of protein processing / dorsal/ventral neural tube patterning / cAMP-dependent protein kinase activity / cellular response to parathyroid hormone stimulus / protein localization to lipid droplet / regulation of bicellular tight junction assembly / cAMP-dependent protein kinase complex / interleukin-2-mediated signaling pathway / sperm capacitation / regulation of osteoblast differentiation / cellular response to cold / protein kinase A regulatory subunit binding / negative regulation of glycolytic process through fructose-6-phosphate / ciliary base / protein kinase A catalytic subunit binding / intracellular potassium ion homeostasis / TORC1 signaling / mesoderm formation / plasma membrane raft / axoneme / sperm flagellum / postsynaptic modulation of chemical synaptic transmission / negative regulation of cAMP/PKA signal transduction / negative regulation of TORC1 signaling / protein serine/threonine/tyrosine kinase activity / protein export from nucleus / cellular response to glucagon stimulus / cellular response to nutrient levels / positive regulation of gluconeogenesis / positive regulation of phagocytosis / acrosomal vesicle / regulation of proteasomal protein catabolic process / positive regulation of protein export from nucleus / neural tube closure / cellular response to glucose stimulus / negative regulation of smoothened signaling pathway / neuromuscular junction / positive regulation of cholesterol biosynthetic process / mRNA processing / positive regulation of insulin secretion / adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway / manganese ion binding / cellular response to heat / adenylate cyclase-activating G protein-coupled receptor signaling pathway / postsynapse / regulation of cell cycle / nuclear speck / protein domain specific binding / protein serine kinase activity / protein serine/threonine kinase activity / centrosome / ubiquitin protein ligase binding / protein kinase binding / perinuclear region of cytoplasm / glutamatergic synapse / magnesium ion binding / mitochondrion / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |   Cricetulus griseus (Chinese hamster) Cricetulus griseus (Chinese hamster) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.833 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.833 Å | ||||||
 Authors Authors | Siefker, C. / Heine, A. / Klebe, G. | ||||||
 Citation Citation | Journal: To Be Published Title: A crystallographic fragment study with cAMP-dependent protein kinase A Authors: Siefker, C. / Heine, A. / Klebe, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5n32.cif.gz | 226.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5n32.ent.gz | 184.7 KB | Display | PDB format |
| PDBx/mmJSON format | 5n32.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n3/5n32ftp://data.pdbj.org/pub/pdb/validation_reports/n3/5n32 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5mhiC  5n1dC  5n1eC  5n1fC  5n1gC  5n1hC  5n1kC  5n1lC  5n1mC  5n1nC  5n1oC  5n36C  5n37C  5n39C  5n3aC  5n3bC  5n3fC  5n3gC  5n3iC  5n3kC  5n3lC  5n3mC  5n3nC  5n3oC  5n3pC  5n3rC  5n3tC  5n7pC  4wihS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41113.766 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: Phosphorylation of S11, T198 and S339 / Source: (gene. exp.) Cricetulus griseus (Chinese hamster) / Gene: PRKACA / Plasmid: pET-16bTEV / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 1993.318 Da / Num. of mol.: 1 / Fragment: UNP residues 12-30 / Source method: obtained synthetically / Details: cAMP dependent protein kinase inhibitor peptide / Source: (synth.) Cricetulus griseus (Chinese hamster) / References: UniProt: A0A061IH64, UniProt: G3HK48*PLUS | ||||||
| #3: Chemical | 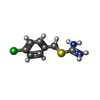  Mass: 201.696 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10ClN2S Mass: 201.696 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H10ClN2S#4: Chemical | ChemComp-MPD / ( |   Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 157 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.83 Å3/Da / Density % sol: 57 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 6.9 / Details: MES-BIS-TRIS Mega8-Solution DTT EDTA LiCl Methanol |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.918409 Å / Beamline: 14.1 / Wavelength: 0.918409 Å |
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Mar 13, 2016 / Details: Silicon, active surface |
| Radiation | Monochromator: Si-111 crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.918409 Å / Relative weight: 1 |
| Reflection | Resolution: 1.83→50 Å / Num. obs: 41140 / % possible obs: 99.8 % / Redundancy: 6.54 % / CC1/2: 1 / Rrim(I) all: 0.047 / Net I/σ(I): 27.26 |
| Reflection shell | Resolution: 1.83→1.94 Å / Redundancy: 6.31 % / Mean I/σ(I) obs: 3.74 / Num. unique obs: 6515 / CC1/2: 0.9 / Rrim(I) all: 0.526 / % possible all: 99.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4WIH Resolution: 1.833→45.548 Å / SU ML: 0.17 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 20.2 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.833→45.548 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
