 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5mzp: Crystal structure of stabilized A2A adenosine receptor A2AR-StaR2... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5mzp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of stabilized A2A adenosine receptor A2AR-StaR2-bRIL in complex with caffeine at 2.1A resolution | ||||||
 Components Components | Adenosine receptor A2a,Soluble cytochrome b562,Adenosine receptor A2a | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / G-PROTEIN-COUPLED RECEPTOR / INTEGRAL MEMBRANE PROTEIN / CHIMERA / THERMOSTABILIZING MUTATIONS | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of norepinephrine secretion / positive regulation of circadian sleep/wake cycle, sleep / Adenosine P1 receptors / positive regulation of acetylcholine secretion, neurotransmission / G protein-coupled adenosine receptor activity / response to purine-containing compound / G protein-coupled adenosine receptor signaling pathway / NGF-independant TRKA activation / Surfactant metabolism / type 5 metabotropic glutamate receptor binding ...regulation of norepinephrine secretion / positive regulation of circadian sleep/wake cycle, sleep / Adenosine P1 receptors / positive regulation of acetylcholine secretion, neurotransmission / G protein-coupled adenosine receptor activity / response to purine-containing compound / G protein-coupled adenosine receptor signaling pathway / NGF-independant TRKA activation / Surfactant metabolism / type 5 metabotropic glutamate receptor binding / negative regulation of vascular permeability / intermediate filament / presynaptic active zone / positive regulation of urine volume / response to caffeine / blood circulation / synaptic transmission, cholinergic / sensory perception / positive regulation of glutamate secretion / eating behavior / regulation of calcium ion transport / alpha-actinin binding / synaptic transmission, dopaminergic / membrane depolarization / asymmetric synapse / axolemma / cellular defense response / prepulse inhibition / phagocytosis / neuron projection morphogenesis / positive regulation of synaptic transmission, glutamatergic / astrocyte activation / presynaptic modulation of chemical synaptic transmission / excitatory postsynaptic potential / regulation of mitochondrial membrane potential / positive regulation of long-term synaptic potentiation / positive regulation of protein secretion / central nervous system development / synaptic transmission, glutamatergic / locomotory behavior / electron transport chain / vasodilation / blood coagulation / adenylate cyclase-modulating G protein-coupled receptor signaling pathway / cell-cell signaling / adenylate cyclase-activating G protein-coupled receptor signaling pathway / presynaptic membrane / negative regulation of neuron apoptotic process / G alpha (s) signalling events / phospholipase C-activating G protein-coupled receptor signaling pathway / positive regulation of ERK1 and ERK2 cascade / calmodulin binding / periplasmic space / electron transfer activity / postsynaptic membrane / response to xenobiotic stimulus / iron ion binding / inflammatory response / negative regulation of cell population proliferation / neuronal cell body / heme binding / apoptotic process / regulation of DNA-templated transcription / lipid binding / dendrite / protein-containing complex binding / glutamatergic synapse / enzyme binding / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Cheng, K.Y.R. / Segala, E. / Robertson, N. / Deflorian, F. / Dore, A.S. / Errey, J.C. / Fiez-Vandal, C. / Marshall, F.H. / Cooke, R.M. | ||||||
 Citation Citation | Journal: Structure / Year: 2017 Title: Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Authors: Cheng, R.K.Y. / Segala, E. / Robertson, N. / Deflorian, F. / Dore, A.S. / Errey, J.C. / Fiez-Vandal, C. / Marshall, F.H. / Cooke, R.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5mzp.cif.gz | 189 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5mzp.ent.gz | 147.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5mzp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mz/5mzpftp://data.pdbj.org/pub/pdb/validation_reports/mz/5mzp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5mzjC  5n2rC  5n2sC  5iu4S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 47996.746 Da / Num. of mol.: 1 Mutation: A54L, T88A, R107A, K122A, N154A, L202A, M1007W, L235A, V239A, S277A, G318A,A54L, T88A, R107A, K122A, N154A, L202A, M1007W, L235A, V239A, S277A, G318A,A54L, T88A, R107A, K122A, N154A, L202A, ...Mutation: A54L, T88A, R107A, K122A, N154A, L202A, M1007W, L235A, V239A, S277A, G318A,A54L, T88A, R107A, K122A, N154A, L202A, M1007W, L235A, V239A, S277A, G318A,A54L, T88A, R107A, K122A, N154A, L202A, M1007W, L235A, V239A, S277A, G318A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human), (gene. exp.) Gene: ADORA2A, ADORA2, cybC / Plasmid: PFAST-BAC / Production host:  Trichoplusia ni (cabbage looper) / Variant (production host): Tni PRO / References: UniProt: P29274, UniProt: P0ABE7 Trichoplusia ni (cabbage looper) / Variant (production host): Tni PRO / References: UniProt: P29274, UniProt: P0ABE7 |
|---|
-Non-polymers , 7 types, 209 molecules 

| #2: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na | ||||||||
|---|---|---|---|---|---|---|---|---|---|
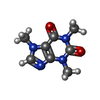
| #3: Chemical | ChemComp-CFF /  Mass: 194.191 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10N4O2 / Comment: medication*YM Mass: 194.191 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10N4O2 / Comment: medication*YM | ||||||||
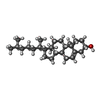
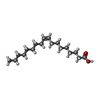
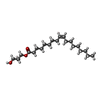
| #4: Chemical | ChemComp-CLR /  Mass: 386.654 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H46O Mass: 386.654 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H46O#5: Chemical | ChemComp-OLA /  Mass: 282.461 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: C18H34O2 Mass: 282.461 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: C18H34O2#6: Chemical | ChemComp-OLC / (  Mass: 356.540 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C21H40O4 Mass: 356.540 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C21H40O4#7: Chemical | ChemComp-TAR / |  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 177 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.58 Å3/Da / Density % sol: 52.36 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: lipidic cubic phase / pH: 5.5 Details: 0.1M MES PH 5.5, 0.2M K/NA TARTRATE, 27.5-40% PEG400, 0.5-1% (V/V) (+/-)-2-METHYL-2,4-PENTANEDIOL |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I24 / Wavelength: 0.96862 Å / Beamline: I24 / Wavelength: 0.96862 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Mar 25, 2016 / Details: (DOUBLE) KB MIRROR PAIR |
| Radiation | Monochromator: DOUBLE SI (111) CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.96862 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→89.94 Å / Num. obs: 29678 / % possible obs: 98.7 % / Redundancy: 4.8 % / CC1/2: 0.996 / Rmerge(I) obs: 0.13 / Net I/σ(I): 8.2 |
| Reflection shell | Resolution: 2.1→2.16 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.977 / Mean I/σ(I) obs: 1.4 / Num. unique obs: 2379 / CC1/2: 0.454 / Rpim(I) all: 0.532 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5IU4 Resolution: 2.1→38.527 Å / SU ML: 0.21 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 21.33
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→38.527 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | S33: -0 Å ° / Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
