 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5lka: Crystal structure of haloalkane dehalogenase LinB 140A+143L+177W+... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5lka | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of haloalkane dehalogenase LinB 140A+143L+177W+211L mutant (LinB86) from Sphingobium japonicum UT26 at 1.3 A resolution | ||||||
 Components Components | Haloalkane dehalogenase | ||||||
 Keywords Keywords | HYDROLASE / haloalkane dehalogenase / bacterial enzyme / mutant | ||||||
| Function / homology |  Function and homology information Function and homology informationhaloalkane dehalogenase / haloalkane dehalogenase activity / response to toxic substance / periplasmic space Similarity search - Function | ||||||
| Biological species |  Sphingobium japonicum (bacteria) Sphingobium japonicum (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.298 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.298 Å | ||||||
 Authors Authors | Degtjarik, O. / Rezacova, P. / Iermak, I. / Chaloupkova, R. / Damborsky, J. / Kuta Smatanova, I. | ||||||
 Citation Citation | Journal: Acs Catalysis / Year: 2016 Title: Engineering a de novo transport tunnel. Authors: Brezovsky, J. / Babkova, P. / Degtjarik, O. / Fortova, A. / Gora, A. / Iermak, I. / Rezacova, P. / Dvorak, P. / Kuta Smatanova, I. / Prokop, Z. / Chaloupkova, R. / Damborsky, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5lka.cif.gz | 151.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5lka.ent.gz | 118.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5lka.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5lka_validation.pdf.gz | 419.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5lka_full_validation.pdf.gz | 419.9 KB | Display | |
| Data in XML | 5lka_validation.xml.gz | 17 KB | Display | |
| Data in CIF | 5lka_validation.cif.gz | 27 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lk/5lkaftp://data.pdbj.org/pub/pdb/validation_reports/lk/5lka | HTTPS FTP |
-Related structure data
| Related structure data |  4wdrS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 33766.184 Da / Num. of mol.: 1 / Mutation: W140A, F143L, L177W, I211L Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sphingobium japonicum (bacteria) / Gene: linB, dhaA, SJA_C1-19590 / Production host: |
|---|---|
| #2: Chemical | ChemComp-SCN / 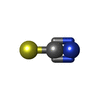  Mass: 58.082 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CNS |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 434 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 434 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.91 Å3/Da / Density % sol: 35.71 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / Details: 0.2 M sodium thiocyanate, 20 % (w/v) PEG 3350. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.9184 Å / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jan 31, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 1.298→46.87 Å / Num. obs: 64480 / % possible obs: 99.6 % / Redundancy: 6.35 % / Biso Wilson estimate: 14.49 Å2 / Rmerge(I) obs: 0.083 / Net I/σ(I): 16.45 |
| Reflection shell | Resolution: 1.298→1.38 Å / Rmerge(I) obs: 0.506 / Mean I/σ(I) obs: 3.5 / % possible all: 98.7 |
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4WDR Resolution: 1.298→40.522 Å / SU ML: 0.12 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 16.13
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 86.72 Å2 / Biso mean: 12.6853 Å2 / Biso min: 4.03 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.298→40.522 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 15
|
