Entry Database : PDB / ID : 5lfaTitle Crystal structure of iron-sulfur cluster containing bacterial (6-4) photolyase PhrB - Y424F mutant with impaired DNA repair activity (6-4) photolyase Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Agrobacterium fabrum (bacteria)Method / / / Resolution : 2.5 Å Authors Kwiatkowski, D. / Zhang, F. / Krauss, N. / Lamparter, T. / Scheerer, P. Funding support Organization Grant number Country German Research Foundation SFB 740 - B6 German Research Foundation SFB 1078 - B6 German Research Foundation Cluster of Excellence - Unifying Concepts in Catalysis - D3/E3
Journal : Photochem. Photobiol. / Year : 2017Title : Crystal Structures of Bacterial (6-4) Photolyase Mutants with Impaired DNA Repair Activity.Authors : Zhang, F. / Ma, H. / Bowatte, K. / Kwiatkowski, D. / Mittmann, E. / Qasem, H. / Krau, N. / Zeng, X. / Ren, Z. / Scheerer, P. / Yang, X. / Lamparter, T. History Deposition Jun 30, 2016 Deposition site / Processing site Revision 1.0 Jan 11, 2017 Provider / Type Revision 1.1 Mar 1, 2017 Group Revision 1.2 Jan 10, 2024 Group Author supporting evidence / Data collection ... Author supporting evidence / Data collection / Database references / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_audit_support / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession / _pdbx_audit_support.funding_organization
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Agrobacterium fabrum (bacteria)
Agrobacterium fabrum (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 3items
Germany, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj
 Assembly
Assembly

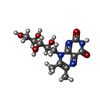



 Mass: 785.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM
Mass: 785.550 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 326.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H18N4O6
Mass: 326.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H18N4O6 Mass: 351.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S4
Mass: 351.640 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe4S4 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.96863 Å
/ Beamline: ID29 / Wavelength: 0.96863 Å Processing
Processing