Entry Database : PDB / ID : 5k8fTitle Crystal structure of Acetyl-CoA Synthetase in complex with ATP and Acetyl-AMP from Cryptococcus neoformans H99 Acetyl-coenzyme A synthetase Keywords / / / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Cryptococcus neoformans var. grubii serotype A (fungus)Method / / / Resolution : 2.45 Å Authors Seattle Structural Genomics Center for Infectious Disease (SSGCID) / Fox III, D. / Delker, S.L. / Potts, K.T. / Lorimer, D.D. / Edwards, T.E. / Mutz, M.W. Journal : To Be Published Title : Crystal structure of Acetyl-CoA Synthetase in complex with ATP and Acetyl-AMP from Cryptococcus neoformans H99Authors : Seattle Structural Genomics Center for Infectious Disease (SSGCID) / Fox III, D. / Delker, S.L. / Potts, K.T. / Numa, M.M. / Edwards, T.E. / Lorimer, D.D. / Mutz, M.W. / Krysan, D.J. History Deposition May 30, 2016 Deposition site / Processing site Revision 1.0 Jul 13, 2016 Provider / Type Revision 2.0 Apr 28, 2021 Group Advisory / Atomic model ... Advisory / Atomic model / Author supporting evidence / Data collection / Database references / Derived calculations / Refinement description / Source and taxonomy / Structure summary Category atom_site / atom_site_anisotrop ... atom_site / atom_site_anisotrop / audit_author / citation_author / diffrn / entity / entity_src_gen / pdbx_entity_instance_feature / pdbx_entity_nonpoly / pdbx_entry_details / pdbx_nonpoly_scheme / pdbx_prerelease_seq / pdbx_refine_tls / pdbx_refine_tls_group / pdbx_struct_assembly / pdbx_struct_assembly_gen / pdbx_struct_conn_angle / pdbx_struct_oper_list / pdbx_struct_sheet_hbond / pdbx_unobs_or_zero_occ_atoms / pdbx_validate_close_contact / pdbx_validate_peptide_omega / pdbx_validate_torsion / refine / refine_hist / refine_ls_restr / refine_ls_shell / reflns / reflns_shell / software / struct_asym / struct_conf / struct_conn / struct_mon_prot_cis / struct_ref_seq_dif / struct_sheet_range / struct_site / struct_site_gen Item _diffrn.pdbx_serial_crystal_experiment / _entity.formula_weight ... _diffrn.pdbx_serial_crystal_experiment / _entity.formula_weight / _entity.pdbx_description / _entity.pdbx_number_of_molecules / _entity_src_gen.gene_src_common_name / _entity_src_gen.pdbx_host_org_scientific_name / _entity_src_gen.pdbx_host_org_strain / _pdbx_entity_nonpoly.comp_id / _pdbx_entity_nonpoly.name / _pdbx_refine_tls.L[1][1] / _pdbx_refine_tls.L[1][2] / _pdbx_refine_tls.L[1][3] / _pdbx_refine_tls.L[2][2] / _pdbx_refine_tls.L[2][3] / _pdbx_refine_tls.L[3][3] / _pdbx_refine_tls.S[1][1] / _pdbx_refine_tls.S[1][2] / _pdbx_refine_tls.S[1][3] / _pdbx_refine_tls.S[2][1] / _pdbx_refine_tls.S[2][2] / _pdbx_refine_tls.S[2][3] / _pdbx_refine_tls.S[3][1] / _pdbx_refine_tls.S[3][2] / _pdbx_refine_tls.S[3][3] / _pdbx_refine_tls.T[1][1] / _pdbx_refine_tls.T[1][2] / _pdbx_refine_tls.T[1][3] / _pdbx_refine_tls.T[2][2] / _pdbx_refine_tls.T[2][3] / _pdbx_refine_tls.T[3][3] / _pdbx_refine_tls.origin_x / _pdbx_refine_tls.origin_y / _pdbx_refine_tls.origin_z / _pdbx_struct_assembly.details / _pdbx_struct_assembly.method_details / _pdbx_struct_assembly_gen.asym_id_list / _pdbx_struct_oper_list.symmetry_operation / _pdbx_struct_sheet_hbond.range_1_auth_comp_id / _pdbx_struct_sheet_hbond.range_1_auth_seq_id / _pdbx_struct_sheet_hbond.range_1_label_comp_id / _pdbx_struct_sheet_hbond.range_1_label_seq_id / _pdbx_struct_sheet_hbond.range_2_auth_comp_id / _pdbx_struct_sheet_hbond.range_2_auth_seq_id / _pdbx_struct_sheet_hbond.range_2_label_comp_id / _pdbx_struct_sheet_hbond.range_2_label_seq_id / _refine.B_iso_max / _refine.B_iso_mean / _refine.B_iso_min / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.ls_number_reflns_R_work / _refine.ls_number_reflns_obs / _refine.ls_percent_reflns_obs / _refine.pdbx_ls_sigma_F / _refine.pdbx_overall_phase_error / _refine.pdbx_stereochemistry_target_values / _refine.solvent_model_details / _refine_hist.cycle_id / _refine_hist.number_atoms_solvent / _refine_hist.number_atoms_total / _refine_hist.pdbx_B_iso_mean_ligand / _refine_hist.pdbx_B_iso_mean_solvent / _refine_hist.pdbx_number_atoms_ligand / _refine_hist.pdbx_number_atoms_protein / _refine_hist.pdbx_number_residues_total / _refine_ls_restr.dev_ideal / _refine_ls_restr.number / _reflns.B_iso_Wilson_estimate / _reflns.pdbx_CC_half / _reflns.pdbx_Rrim_I_all / _reflns.pdbx_chi_squared / _reflns.pdbx_redundancy / _struct_conf.beg_auth_asym_id / _struct_conf.beg_auth_comp_id / _struct_conf.beg_auth_seq_id / _struct_conf.beg_label_asym_id / _struct_conf.beg_label_comp_id / _struct_conf.beg_label_seq_id / _struct_conf.end_auth_asym_id / _struct_conf.end_auth_comp_id / _struct_conf.end_auth_seq_id / _struct_conf.end_label_asym_id / _struct_conf.end_label_comp_id / _struct_conf.end_label_seq_id / _struct_conf.pdbx_PDB_helix_class / _struct_conf.pdbx_PDB_helix_length / _struct_mon_prot_cis.pdbx_omega_angle / _struct_ref_seq_dif.details / _struct_sheet_range.beg_auth_comp_id / _struct_sheet_range.beg_auth_seq_id / _struct_sheet_range.beg_label_comp_id / _struct_sheet_range.beg_label_seq_id / _struct_sheet_range.end_auth_comp_id / _struct_sheet_range.end_auth_seq_id / _struct_sheet_range.end_label_comp_id / _struct_sheet_range.end_label_seq_id Description Details Provider / Type Revision 2.1 Sep 27, 2023 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Cryptococcus neoformans var. grubii serotype A (fungus)
Cryptococcus neoformans var. grubii serotype A (fungus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly





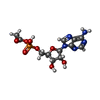




 Mass: 507.181 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Feature type: SUBJECT OF INVESTIGATION / Comment: ATP, energy-carrying molecule*YM
Mass: 507.181 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Feature type: SUBJECT OF INVESTIGATION / Comment: ATP, energy-carrying molecule*YM Mass: 389.258 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H16N5O8P / Feature type: SUBJECT OF INVESTIGATION
Mass: 389.258 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H16N5O8P / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 62.068 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Sample preparation
Sample preparation / Beamline: 21-ID-G / Wavelength: 0.97856 Å
/ Beamline: 21-ID-G / Wavelength: 0.97856 Å Processing
Processing