 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5jz6: Aspartyl/Asparaginyl beta-hydroxylase (AspH)oxygenase and TPR dom... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5jz6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Aspartyl/Asparaginyl beta-hydroxylase (AspH)oxygenase and TPR domains in complex with manganese and L-malate | ||||||
 Components Components | Aspartyl/asparaginyl beta-hydroxylase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / 2-oxoglutarate dependent oxygenase / aspartyl/asparaginyl beta-hydroxylase / EGF-like domain hydroxylase / double stranded beta-helix / tetratricopeptide repeat | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptide-aspartate beta-dioxygenase / regulation of protein depolymerization / activation of store-operated calcium channel activity / regulation of cell communication by electrical coupling / junctional sarcoplasmic reticulum membrane / peptidyl-aspartic acid 3-dioxygenase activity / sarcoplasmic reticulum lumen / cortical endoplasmic reticulum / positive regulation of intracellular protein transport / structural constituent of muscle ...peptide-aspartate beta-dioxygenase / regulation of protein depolymerization / activation of store-operated calcium channel activity / regulation of cell communication by electrical coupling / junctional sarcoplasmic reticulum membrane / peptidyl-aspartic acid 3-dioxygenase activity / sarcoplasmic reticulum lumen / cortical endoplasmic reticulum / positive regulation of intracellular protein transport / structural constituent of muscle / response to ATP / positive regulation of proteolysis / phosphatidylinositol-mediated signaling / positive regulation of calcium ion transport into cytosol / Protein hydroxylation / detection of calcium ion / calcium ion homeostasis / regulation of cytosolic calcium ion concentration / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / Ion homeostasis / regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ion / muscle contraction / sarcoplasmic reticulum membrane / calcium channel complex / cellular response to calcium ion / regulation of protein stability / Stimuli-sensing channels / calcium ion transmembrane transport / transmembrane transporter binding / electron transfer activity / calcium ion binding / positive regulation of DNA-templated transcription / endoplasmic reticulum membrane / structural molecule activity / endoplasmic reticulum / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.354 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.354 Å | ||||||
 Authors Authors | McDonough, M.A. / Pfeffer, I. | ||||||
 Citation Citation | Journal: Nat Commun / Year: 2019 Title: Aspartate/asparagine-beta-hydroxylase crystal structures reveal an unexpected epidermal growth factor-like domain substrate disulfide pattern. Authors: Pfeffer, I. / Brewitz, L. / Krojer, T. / Jensen, S.A. / Kochan, G.T. / Kershaw, N.J. / Hewitson, K.S. / McNeill, L.A. / Kramer, H. / Munzel, M. / Hopkinson, R.J. / Oppermann, U. / Handford, ...Authors: Pfeffer, I. / Brewitz, L. / Krojer, T. / Jensen, S.A. / Kochan, G.T. / Kershaw, N.J. / Hewitson, K.S. / McNeill, L.A. / Kramer, H. / Munzel, M. / Hopkinson, R.J. / Oppermann, U. / Handford, P.A. / McDonough, M.A. / Schofield, C.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5jz6.cif.gz | 194.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5jz6.ent.gz | 153.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5jz6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jz/5jz6ftp://data.pdbj.org/pub/pdb/validation_reports/jz/5jz6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5apaC  5jqyC  5jz8C  5jzaC  5jzuC  6rk9C C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 49405.336 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ASPH, BAH / Production host:  References: UniProt: Q12797, peptide-aspartate beta-dioxygenase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mn | ||||||
| #3: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical | ChemComp-LMR / ( | 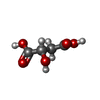  Mass: 134.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O5 Mass: 134.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O5#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.02 Å3/Da / Density % sol: 59.32 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 1mM MMT buffer (DL-Malic acid, MES, Tris base), 25% PEG1500, 1 mM MnCl2, 2 mM N-oxalylglycine, 18 mg/ml protein |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 0.92 Å / Beamline: I04 / Wavelength: 0.92 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Jun 10, 2014 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 2.35→47.16 Å / Num. obs: 25633 / % possible obs: 99.8 % / Redundancy: 6.5 % / Biso Wilson estimate: 35.39 Å2 / Rmerge(I) obs: 0.135 / Net I/σ(I): 14.2 |
| Reflection shell | Resolution: 2.35→2.43 Å / Redundancy: 5.9 % / Rmerge(I) obs: 0.896 / Mean I/σ(I) obs: 2.5 / % possible all: 99.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.354→47.153 Å / SU ML: 0.25 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 20.53
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 165.42 Å2 / Biso mean: 51.3736 Å2 / Biso min: 22.33 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.354→47.153 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 14
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
