 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5jui: domain-swapped dimer of the the KRT10-binding region (BR) of PsrP -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5jui | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | domain-swapped dimer of the the KRT10-binding region (BR) of PsrP | |||||||||||||||
 Components Components | Cell wall surface anchor family protein | |||||||||||||||
 Keywords Keywords | STRUCTURAL PROTEIN / Streptococcus pneumoniae / Pneumococcal Serine Rich Repeat Protein / Oligomerisation / Bacterial aggregation / Biofilm formation / DNA | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationGram-positive-bacterium-type cell wall / single-species biofilm formation / symbiont-mediated perturbation of host defense response / cell adhesion / cell surface / DNA binding Similarity search - Function | |||||||||||||||
| Biological species |  Streptococcus pneumoniae serotype 4 (bacteria) Streptococcus pneumoniae serotype 4 (bacteria) | |||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | |||||||||||||||
 Authors Authors | Schulte, T. / Mikaelsson, C. / Achour, A. | |||||||||||||||
| Funding support |  Sweden, 4items Sweden, 4items
| |||||||||||||||
 Citation Citation | Journal: Sci Rep / Year: 2016 Title: The BR domain of PsrP interacts with extracellular DNA to promote bacterial aggregation; structural insights into pneumococcal biofilm formation. Authors: Tim Schulte / Cecilia Mikaelsson / Audrey Beaussart / Alexey Kikhney / Maya Deshmukh / Sebastian Wolniak / Anuj Pathak / Christine Ebel / Jonas Löfling / Federico Fogolari / Birgitta ...Authors: Tim Schulte / Cecilia Mikaelsson / Audrey Beaussart / Alexey Kikhney / Maya Deshmukh / Sebastian Wolniak / Anuj Pathak / Christine Ebel / Jonas Löfling / Federico Fogolari / Birgitta Henriques-Normark / Yves F Dufrêne / Dmitri Svergun / Per-Åke Nygren / Adnane Achour /     Abstract: The major human pathogen Streptococcus pneumoniae is a leading cause of disease and death worldwide. Pneumococcal biofilm formation within the nasopharynx leads to long-term colonization and ...The major human pathogen Streptococcus pneumoniae is a leading cause of disease and death worldwide. Pneumococcal biofilm formation within the nasopharynx leads to long-term colonization and persistence within the host. We have previously demonstrated that the capsular surface-associated pneumococcal serine rich repeat protein (PsrP), key factor for biofilm formation, binds to keratin-10 (KRT10) through its microbial surface component recognizing adhesive matrix molecule (MSCRAMM)-related globular binding region domain (BR187-385). Here, we show that BR187-385 also binds to DNA, as demonstrated by electrophoretic mobility shift assays and size exclusion chromatography. Further, heterologous expression of BR187-378 or the longer BR120-378 construct on the surface of a Gram-positive model host bacterium resulted in the formation of cellular aggregates that was significantly enhanced in the presence of DNA. Crystal structure analyses revealed the formation of BR187-385 homo-dimers via an intermolecular β-sheet, resulting in a positively charged concave surface, shaped to accommodate the acidic helical DNA structure. Furthermore, small angle X-ray scattering and circular dichroism studies indicate that the aggregate-enhancing N-terminal region of BR120-166 adopts an extended, non-globular structure. Altogether, our results suggest that PsrP adheres to extracellular DNA in the biofilm matrix and thus promotes pneumococcal biofilm formation. #1: Journal: Open Biol / Year: 2014Title: The basic keratin 10-binding domain of the virulence-associated pneumococcal serine-rich protein PsrP adopts a novel MSCRAMM fold. Authors: Tim Schulte / Jonas Löfling / Cecilia Mikaelsson / Alexey Kikhney / Karina Hentrich / Aurora Diamante / Christine Ebel / Staffan Normark / Dmitri Svergun / Birgitta Henriques-Normark / Adnane Achour / Abstract: Streptococcus pneumoniae is a major human pathogen, and a leading cause of disease and death worldwide. Pneumococcal invasive disease is triggered by initial asymptomatic colonization of the human ...Streptococcus pneumoniae is a major human pathogen, and a leading cause of disease and death worldwide. Pneumococcal invasive disease is triggered by initial asymptomatic colonization of the human upper respiratory tract. The pneumococcal serine-rich repeat protein (PsrP) is a lung-specific virulence factor whose functional binding region (BR) binds to keratin-10 (KRT10) and promotes pneumococcal biofilm formation through self-oligomerization. We present the crystal structure of the KRT10-binding domain of PsrP (BR187-385) determined to 2.0 Å resolution. BR187-385 adopts a novel variant of the DEv-IgG fold, typical for microbial surface components recognizing adhesive matrix molecules adhesins, despite very low sequence identity. An extended β-sheet on one side of the compressed, two-sided barrel presents a basic groove that possibly binds to the acidic helical rod domain of KRT10. Our study also demonstrates the importance of the other side of the barrel, formed by extensive well-ordered loops and stabilized by short β-strands, for interaction with KRT10. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5jui.cif.gz | 223 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5jui.ent.gz | 180.8 KB | Display | PDB format |
| PDBx/mmJSON format | 5jui.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ju/5juiftp://data.pdbj.org/pub/pdb/validation_reports/ju/5jui | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3zghS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 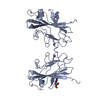
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 21407.662 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus pneumoniae serotype 4 (strain ATCC BAA-334 / TIGR4) (bacteria)Strain: ATCC BAA-334 / TIGR4 / Gene: SP_1772 / Production host: #2: Chemical | ChemComp-NA / |   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#3: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.54 Å3/Da / Density % sol: 65.29 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion Details: The BR187-378 dimer was concentrated to 20 mg/mL in 20 mM Na-Citrate, 500 mM NaCl, 10% (V/V) glycerol, pH 5.5. Well-diffracting crystals were obtained in 100 mM Trisodiumcitrate dihydrate pH ...Details: The BR187-378 dimer was concentrated to 20 mg/mL in 20 mM Na-Citrate, 500 mM NaCl, 10% (V/V) glycerol, pH 5.5. Well-diffracting crystals were obtained in 100 mM Trisodiumcitrate dihydrate pH 5.6; 34-44% 2-methyl-4-pentanediole using the sitting drop vapor-diffusion method. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF / Beamline: ID23-1 / Wavelength: 0.95376 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Sep 28, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95376 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→48.4 Å / Num. obs: 53483 / % possible obs: 100 % / Observed criterion σ(I): 2 / Redundancy: 6.6 % / Biso Wilson estimate: 49.1 Å2 / CC1/2: 1 / Rsym value: 0.059 / Net I/σ(I): 16.1 |
| Reflection shell | Resolution: 2.1→2.18 Å / Redundancy: 6.4 % / Rmerge(I) obs: 0.864 / Mean I/σ(I) obs: 2 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3ZGH Resolution: 2.1→48.361 Å / SU ML: 0.26 / Cross valid method: FREE R-VALUE / σ(F): 1.33 / Phase error: 25.97 Details: A molecular replacement search performed using Phaser revealed three molecules of the BR187-385 monomer (PDB: 3ZGH) within the asymmetric unit. Initial rigid body and restrained refinement ...Details: A molecular replacement search performed using Phaser revealed three molecules of the BR187-385 monomer (PDB: 3ZGH) within the asymmetric unit. Initial rigid body and restrained refinement rounds were performed in CCP4 refmac, and Coot was used for manual model rebuilding. The mFo-DFc electron density difference map clearly indicated that residues Q312-G315 had to be re-built such that residues L202-G315 of chain A were linked to residues Y316-S377 of the symmetry mate [X, -Y, -Z + (1 0 0) & {-1 0 0}), symmetry operation in Coot], and vice versa. The same procedure was applied to swap the corresponding regions between chains B and C. The model was thereafter refined using Phenix with individual isotropic ADP factors, TLS refinement and NCS torsion restraints. The final model was refined to R and Rfree values of 21.1 and 24.4, respectively, and comprised residues L202-S377, residues N203-S377 and residues N203-S376 for chains A, B and C, respectively. The electron density for the structural model of chain A was of higher quality compared to the density for both chains B and C, which was corroborated by higher values for real-space correlation coefficients. This difference in the quality of the electron density map was also true for the crucial loop region around residue S314.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→48.361 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|