 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5jib: Crystal structure of the Thermotoga maritima acetyl esterase (TM0... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5jib | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the Thermotoga maritima acetyl esterase (TM0077) complex with a substrate analog | ||||||
 Components Components | Cephalosporin-C deacetylase | ||||||
 Keywords Keywords | HYDROLASE / CARBOHYDRATE METABOLISM / CEPHALOSPORIN DEACETYLASE / ROSSMANN FOLD | ||||||
| Function / homology |  Function and homology information Function and homology informationxylan metabolic process / cephalosporin C metabolic process / cephalosporin-C deacetylase / cephalosporin-C deacetylase activity / acetylxylan esterase / polysaccharide metabolic process / acetylxylan esterase activity / carboxylic ester hydrolase activity / cellulose catabolic process / calcium ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   Thermotoga maritima (bacteria) Thermotoga maritima (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.86 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.86 Å | ||||||
 Authors Authors | Manoj, N. | ||||||
| Funding support |  India, 1items India, 1items
| ||||||
 Citation Citation | Journal: Biochem. Biophys. Res. Commun. / Year: 2016 Title: Crystal structure of Thermotoga maritima acetyl esterase complex with a substrate analog: Insights into the distinctive substrate specificity in the CE7 carbohydrate esterase family Authors: Singh, M.K. / Manoj, N. #1: Journal: J. Struct. Biol. / Year: 2016Title: An extended loop in CE7 carbohydrate esterase family is dispensable for oligomerization but required for activity and thermostability. Authors: Singh, M.K. / Manoj, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5jib.cif.gz | 400.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5jib.ent.gz | 322.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5jib.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ji/5jibftp://data.pdbj.org/pub/pdb/validation_reports/ji/5jib | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5fdfS S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 38662.922 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima (strain ATCC 43589 / MSB8 / DSM 3109 / JCM 10099) (bacteria)Strain: ATCC 43589 / MSB8 / DSM 3109 / JCM 10099 / Gene: axeA, TM_0077 / Plasmid: PMH1 / Production host: References: UniProt: Q9WXT2, cephalosporin-C deacetylase, acetylxylan esterase #2: Chemical | ChemComp-OIA / [( 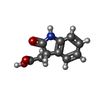  Mass: 191.183 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H9NO3 Mass: 191.183 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H9NO3#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1039 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1039 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.41 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 0.05 M ammonium sulfate, 0.1 M Bis-Tris pH 6.5 and 30% (V/V) pentaerythritolethoxylate (15/4 EO/OH) |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 27, 2014 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.86→45.97 Å / Num. obs: 156655 / % possible obs: 94.3 % / Redundancy: 7.5 % / CC1/2: 0.998 / Rmerge(I) obs: 0.072 / Rpim(I) all: 0.028 / Rrim(I) all: 0.077 / Net I/σ(I): 19 / Num. measured all: 1178812 / Scaling rejects: 323 |
| Reflection shell | Resolution: 1.86→1.89 Å / Redundancy: 6.1 % / Rmerge(I) obs: 0.36 / Mean I/σ(I) obs: 5.1 / % possible all: 70.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5FDF Resolution: 1.86→45.97 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.944 / SU B: 2.508 / SU ML: 0.076 / SU R Cruickshank DPI: 0.117 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.132 / ESU R Free: 0.122 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 64.16 Å2 / Biso mean: 19.604 Å2 / Biso min: 8.62 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.175 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.86→45.97 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.863→1.912 Å / Total num. of bins used: 20
|
