 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5j5k | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF AFMP4P IN COMPLEX WITH PALMITIC ACID | ||||||
 Components Components | Uncharacterized protein | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / VIRULENCE FACTOR / AFMP4 / PALMITIC ACID | ||||||
| Function / homology | Cell wall mannoprotein 1 / Hydrophobic surface binding protein A / fungal-type cell wall / lipid binding / extracellular region / alpha-D-mannopyranose / PALMITIC ACID / Cell wall mannoprotein 1 Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | ||||||
 Authors Authors | Zhang, H. / Hao, Q. | ||||||
 Citation Citation | Journal: To Be Published Title: A Novel Class Of Virulence Factors In Penicillium Marneffei And Aspergillus Fumigatus Enhances Intracellular Survival In Monocytes By Arachidonic Acid Binding Authors: Zhang, H. / Hao, Q. / Lam, W.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5j5k.cif.gz | 73.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5j5k.ent.gz | 54.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5j5k.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5j5k_validation.pdf.gz | 560.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5j5k_full_validation.pdf.gz | 560.5 KB | Display | |
| Data in XML | 5j5k_validation.xml.gz | 4.8 KB | Display | |
| Data in CIF | 5j5k_validation.cif.gz | 7.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j5/5j5kftp://data.pdbj.org/pub/pdb/validation_reports/j5/5j5k | HTTPS FTP |
-Related structure data
| Related structure data |  5j5lC  3l1nS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 16915.633 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 38-194 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Strain: ATCC MYA-4609 / Af293 / CBS 101355 / FGSC A1100 / Gene: AFUA_2G17630 / Plasmid: PPIC9K / Production host: PICHIA PASTORIS (fungus) / Strain (production host): X33 / References: UniProt: Q4WZA5 |
|---|---|
| #2: Sugar | ChemComp-MAN / 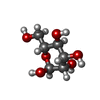  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
| #3: Chemical | ChemComp-PLM /   Mass: 256.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H32O2 Mass: 256.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H32O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 148 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48.66 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 0.1M MES PH 6.5, 25% POLYETHYLENE GLYCOL 4000, 0.2M MGCL2, VAPOR DIFFUSION, HANGING DROP, TEMPERATURE 298K PH range: 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF  / Beamline: BL17U / Wavelength: 0.97922 Å / Beamline: BL17U / Wavelength: 0.97922 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jun 10, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97922 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→50 Å / Num. obs: 12425 / % possible obs: 99.8 % / Redundancy: 21.7 % / Rmerge(I) obs: 0.061 / Net I/σ(I): 13.2 |
| Reflection shell | Resolution: 1.95→2.02 Å / Redundancy: 19.9 % / Rmerge(I) obs: 0.433 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3L1N Resolution: 1.95→50 Å / Cor.coef. Fo:Fc: 0.97 / Cor.coef. Fo:Fc free: 0.958 / SU B: 6.354 / SU ML: 0.084 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.127 / ESU R Free: 0.119 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.42 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|