 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5h0h: Crystal structure of HCK complexed with a pyrrolo-pyrimidine inhi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5h0h | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of HCK complexed with a pyrrolo-pyrimidine inhibitor (S)-2-(((1r,4S)-4-(4-amino-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)cyclohexyl)amino)-N,N,4-trimethylpentanamide | ||||||
 Components Components | Tyrosine-protein kinase HCK | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationleukocyte degranulation / innate immune response-activating signaling pathway / respiratory burst after phagocytosis / leukocyte migration involved in immune response / regulation of podosome assembly / FLT3 signaling through SRC family kinases / regulation of phagocytosis / regulation of DNA-binding transcription factor activity / Nef and signal transduction / positive regulation of actin filament polymerization ...leukocyte degranulation / innate immune response-activating signaling pathway / respiratory burst after phagocytosis / leukocyte migration involved in immune response / regulation of podosome assembly / FLT3 signaling through SRC family kinases / regulation of phagocytosis / regulation of DNA-binding transcription factor activity / Nef and signal transduction / positive regulation of actin filament polymerization / Fc-gamma receptor signaling pathway involved in phagocytosis / mesoderm development / FCGR activation / type II interferon-mediated signaling pathway / transport vesicle / Signaling by CSF3 (G-CSF) / phosphotyrosine residue binding / FCGR3A-mediated IL10 synthesis / cell surface receptor protein tyrosine kinase signaling pathway / lipopolysaccharide-mediated signaling pathway / integrin-mediated signaling pathway / cell projection / regulation of actin cytoskeleton organization / FCGR3A-mediated phagocytosis / non-membrane spanning protein tyrosine kinase activity / Regulation of signaling by CBL / non-specific protein-tyrosine kinase / negative regulation of inflammatory response to antigenic stimulus / peptidyl-tyrosine phosphorylation / cytokine-mediated signaling pathway / Inactivation of CSF3 (G-CSF) signaling / caveola / cytoplasmic side of plasma membrane / Signaling by CSF1 (M-CSF) in myeloid cells / regulation of cell shape / protein autophosphorylation / regulation of inflammatory response / protein tyrosine kinase activity / cell differentiation / cytoskeleton / lysosome / cell adhesion / defense response to Gram-positive bacterium / intracellular signal transduction / protein phosphorylation / inflammatory response / signaling receptor binding / focal adhesion / intracellular membrane-bounded organelle / positive regulation of cell population proliferation / lipid binding / negative regulation of apoptotic process / Golgi apparatus / ATP binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.72 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.72 Å | ||||||
 Authors Authors | Tomabechi, Y. / Kukimoto-Niino, M. / Shirouzu, M. | ||||||
 Citation Citation | Journal: Bioorg. Med. Chem. / Year: 2017 Title: Activity cliff for 7-substituted pyrrolo-pyrimidine inhibitors of HCK explained in terms of predicted basicity of the amine nitrogen. Authors: Yuki, H. / Kikuzato, K. / Koda, Y. / Mikuni, J. / Tomabechi, Y. / Kukimoto-Niino, M. / Tanaka, A. / Shirai, F. / Shirouzu, M. / Koyama, H. / Honma, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5h0h.cif.gz | 118 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5h0h.ent.gz | 86 KB | Display | PDB format |
| PDBx/mmJSON format | 5h0h.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5h0h_validation.pdf.gz | 712.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5h0h_full_validation.pdf.gz | 718.8 KB | Display | |
| Data in XML | 5h0h_validation.xml.gz | 21.9 KB | Display | |
| Data in CIF | 5h0h_validation.cif.gz | 33 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h0/5h0hftp://data.pdbj.org/pub/pdb/validation_reports/h0/5h0h | HTTPS FTP |
-Related structure data
| Related structure data |  5h09C  5h0bC  5h0eC  5h0gC  3vs3S  5h0a 5h0c 5h0d 5h0f C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 52000.227 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 81-526 / Mutation: Q523E, Q524E, Q525I Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HCK / Production host:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm)References: UniProt: P08631, non-specific protein-tyrosine kinase |
|---|---|
| #2: Chemical | ChemComp-OOV / (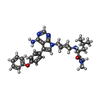  Mass: 540.699 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C32H40N6O2 Mass: 540.699 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C32H40N6O2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 393 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 393 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 46.87 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / Details: 0.22-0.25 M ammonium formate, 12-22 % PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL26B2 / Wavelength: 1 Å / Beamline: BL26B2 / Wavelength: 1 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Jan 23, 2015 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.72→50 Å / Num. obs: 51602 / % possible obs: 99.9 % / Redundancy: 7.3 % / Biso Wilson estimate: 22.17 Å2 / Rmerge(I) obs: 0.068 / Rpim(I) all: 0.027 / Rrim(I) all: 0.073 / Χ2: 1.132 / Net I/av σ(I): 34.653 / Net I/σ(I): 9.3 / Num. measured all: 375872 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3VS3 Resolution: 1.72→42.812 Å / SU ML: 0.18 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 21.81
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 133.98 Å2 / Biso mean: 37.3881 Å2 / Biso min: 11.22 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.72→42.812 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 14
|
