Entry Database : PDB / ID : 5gqjTitle Crystal structure of Cypovirus Polyhedra mutant with deletion of Ser193 and Ala194 Polyhedrin Keywords / / Function / homology / / / / / / Biological species Method / / / Resolution : 1.5 Å Authors Abe, S. / Tabe, H. / Ijiri, H. / Yamashita, K. / Hirata, K. / Mori, H. / Ueno, T. Journal : ACS Nano / Year : 2017Title : Crystal Engineering of Self-Assembled Porous Protein Materials in Living CellsAuthors : Abe, S. / Tabe, H. / Ijiri, H. / Yamashita, K. / Hirata, K. / Atsumi, K. / Shimoi, T. / Akai, M. / Mori, H. / Kitagawa, S. / Ueno, T. History Deposition Aug 7, 2016 Deposition site / Processing site Revision 1.0 Feb 15, 2017 Provider / Type Revision 1.1 Apr 12, 2017 Group Revision 1.2 Sep 27, 2017 Group / Category / diffrn_sourceItem / _diffrn_source.pdbx_synchrotron_siteRevision 1.3 Nov 8, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id Revision 1.4 Oct 9, 2024 Group / Category / pdbx_modification_feature
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Bombyx mori cypovirus 1
Bombyx mori cypovirus 1 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj
 Assembly
Assembly


 Spodoptera frugiperda (fall armyworm) / References: UniProt: P11041
Spodoptera frugiperda (fall armyworm) / References: UniProt: P11041




 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM
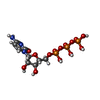
Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 483.156 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H16N3O14P3
Mass: 483.156 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H16N3O14P3 Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O2 Sample preparation
Sample preparation / Beamline: BL41XU / Wavelength: 1 Å
/ Beamline: BL41XU / Wavelength: 1 Å Processing
Processing