 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5gkn: Catalase structure determined by electron crystallography of thin... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5gkn | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Catalase structure determined by electron crystallography of thin 3D crystals | |||||||||
 Components Components | Catalase | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / HEME / NADPH | |||||||||
| Function / homology |  Function and homology information Function and homology informationDetoxification of Reactive Oxygen Species / Peroxisomal protein import / catalase / catalase activity / Neutrophil degranulation / peroxisomal matrix / positive regulation of cell division / hydrogen peroxide catabolic process / response to hydrogen peroxide / peroxisome ...Detoxification of Reactive Oxygen Species / Peroxisomal protein import / catalase / catalase activity / Neutrophil degranulation / peroxisomal matrix / positive regulation of cell division / hydrogen peroxide catabolic process / response to hydrogen peroxide / peroxisome / heme binding / enzyme binding / mitochondrion / metal ion binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | ELECTRON CRYSTALLOGRAPHY / electron crystallography / cryo EM / Resolution: 3.2 Å | |||||||||
 Authors Authors | Yonekura, K. | |||||||||
| Funding support |  Japan, 1items Japan, 1items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2015 Title: Electron crystallography of ultrathin 3D protein crystals: atomic model with charges. Authors: Koji Yonekura / Kazuyuki Kato / Mitsuo Ogasawara / Masahiro Tomita / Chikashi Toyoshima / Abstract: Membrane proteins and macromolecular complexes often yield crystals too small or too thin for even the modern synchrotron X-ray beam. Electron crystallography could provide a powerful means for ...Membrane proteins and macromolecular complexes often yield crystals too small or too thin for even the modern synchrotron X-ray beam. Electron crystallography could provide a powerful means for structure determination with such undersized crystals, as protein atoms diffract electrons four to five orders of magnitude more strongly than they do X-rays. Furthermore, as electron crystallography yields Coulomb potential maps rather than electron density maps, it could provide a unique method to visualize the charged states of amino acid residues and metals. Here we describe an attempt to develop a methodology for electron crystallography of ultrathin (only a few layers thick) 3D protein crystals and present the Coulomb potential maps at 3.4-Å and 3.2-Å resolution, respectively, obtained from Ca(2+)-ATPase and catalase crystals. These maps demonstrate that it is indeed possible to build atomic models from such crystals and even to determine the charged states of amino acid residues in the Ca(2+)-binding sites of Ca(2+)-ATPase and that of the iron atom in the heme in catalase. #1: Journal: Proc Natl Acad Sci U S A / Year: 2015Title: Electron crystallography of ultrathin 3D protein crystals: atomic model with charges. Authors: Koji Yonekura / Kazuyuki Kato / Mitsuo Ogasawara / Masahiro Tomita / Chikashi Toyoshima / Abstract: Membrane proteins and macromolecular complexes often yield crystals too small or too thin for even the modern synchrotron X-ray beam. Electron crystallography could provide a powerful means for ...Membrane proteins and macromolecular complexes often yield crystals too small or too thin for even the modern synchrotron X-ray beam. Electron crystallography could provide a powerful means for structure determination with such undersized crystals, as protein atoms diffract electrons four to five orders of magnitude more strongly than they do X-rays. Furthermore, as electron crystallography yields Coulomb potential maps rather than electron density maps, it could provide a unique method to visualize the charged states of amino acid residues and metals. Here we describe an attempt to develop a methodology for electron crystallography of ultrathin (only a few layers thick) 3D protein crystals and present the Coulomb potential maps at 3.4-Å and 3.2-Å resolution, respectively, obtained from Ca(2+)-ATPase and catalase crystals. These maps demonstrate that it is indeed possible to build atomic models from such crystals and even to determine the charged states of amino acid residues in the Ca(2+)-binding sites of Ca(2+)-ATPase and that of the iron atom in the heme in catalase. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5gkn.cif.gz | 411.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5gkn.ent.gz | 337.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5gkn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gk/5gknftp://data.pdbj.org/pub/pdb/validation_reports/gk/5gkn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3j7tC  3nwlS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 59999.160 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) #2: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4#3: Chemical | ChemComp-NDP / 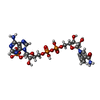  Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON CRYSTALLOGRAPHY / Number of used crystals: 58 |
|---|---|
| EM experiment | Aggregation state: 3D ARRAY / 3D reconstruction method: electron crystallography |
- Sample preparation
Sample preparation
| Component | Name: catalase / Type: COMPLEX / Entity ID: #1 / Source: NATURAL |
|---|---|
| Source (natural) | Organism: |
| Buffer solution | pH: 5.3 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Vitrification | Cryogen name: ETHANE |
| Crystal grow | pH: 5.3 |
-Data collection
| Microscopy | Model: HITACHI EF3000 |
|---|---|
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD |
| Image recording | Electron dose: 0.01 e/Å2 / Film or detector model: TVIPS TEMCAM-F224 (2k x 2k) |
| EM diffraction | Camera length: 3450 mm |
| EM diffraction shell | Resolution: 3.2→20 Å / Fourier space coverage: 73 % / Multiplicity: 46.3 / Num. of structure factors: 30337 / Phase residual: 23.7 ° |
| EM diffraction stats | Fourier space coverage: 73 % / High resolution: 3.2 Å / Num. of intensities measured: 10000 / Num. of structure factors: 30337 / Phase error: 23.7 ° / Phase residual: 23.7 ° / Phase error rejection criteria: unknown / Rmerge: 33.2 / Rsym: 10 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software | Name: PHENIX / Category: model fitting | |||||||||||||||||||||||||||||||||||||||||||||||||
| EM 3D crystal entity | ∠α: 90 ° / ∠β: 90 ° / ∠γ: 90 ° / A: 69 Å / B: 173.5 Å / C: 206 Å / Space group name: P212121 / Space group num: 19 | |||||||||||||||||||||||||||||||||||||||||||||||||
| CTF correction | Type: NONE | |||||||||||||||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Symmetry type: 3D CRYSTAL | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | Starting model: 3NWL Resolution: 3.2→19.988 Å / SU ML: 0.49 / Cross valid method: FREE R-VALUE / σ(F): 1.81 / Phase error: 23.98 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
