 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5f02: CATHEPSIN L IN COMPLEX WITH (2S,4R)-4-(2-Chloro-4-methoxy-benzene... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5f02 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CATHEPSIN L IN COMPLEX WITH (2S,4R)-4-(2-Chloro-4-methoxy-benzenesulfonyl)-1-[3-(5-chloro-pyridin-2-yl)-azetidine-3-carbonyl]-pyrrolidine-2-carboxylic acid (1-cyano-cyclopropyl)-amide | ||||||
 Components Components | Cathepsin L1 | ||||||
 Keywords Keywords | HYDROLASE / PROTEASE INHIBITOR / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationenkephalin processing / cathepsin L / CD4-positive, alpha-beta T cell lineage commitment / macrophage apoptotic process / chromaffin granule / elastin catabolic process / antigen processing and presentation of peptide antigen / RUNX1 regulates transcription of genes involved in differentiation of keratinocytes / endolysosome lumen / cellular response to thyroid hormone stimulus ...enkephalin processing / cathepsin L / CD4-positive, alpha-beta T cell lineage commitment / macrophage apoptotic process / chromaffin granule / elastin catabolic process / antigen processing and presentation of peptide antigen / RUNX1 regulates transcription of genes involved in differentiation of keratinocytes / endolysosome lumen / cellular response to thyroid hormone stimulus / Trafficking and processing of endosomal TLR / zymogen activation / proteoglycan binding / Assembly of collagen fibrils and other multimeric structures / antigen processing and presentation / protein autoprocessing / Collagen degradation / collagen catabolic process / fibronectin binding / serpin family protein binding / collagen binding / Attachment and Entry / receptor-mediated endocytosis of virus by host cell / Degradation of the extracellular matrix / multivesicular body / endocytic vesicle lumen / cysteine-type peptidase activity / MHC class II antigen presentation / lysosomal lumen / proteolysis involved in protein catabolic process / Endosomal/Vacuolar pathway / antigen processing and presentation of exogenous peptide antigen via MHC class II / : / histone binding / adaptive immune response / Attachment and Entry / lysosome / apical plasma membrane / fusion of virus membrane with host plasma membrane / cysteine-type endopeptidase activity / intracellular membrane-bounded organelle / fusion of virus membrane with host endosome membrane / symbiont entry into host cell / Golgi apparatus / proteolysis / extracellular space / extracellular exosome / extracellular region / nucleus / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.43 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.43 Å | ||||||
 Authors Authors | Banner, D. / Benz, J. / Stihle, M. / Kuglstatter, A. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2016 Title: A Real-World Perspective on Molecular Design. Authors: Kuhn, B. / Guba, W. / Hert, J. / Banner, D. / Bissantz, C. / Ceccarelli, S. / Haap, W. / Korner, M. / Kuglstatter, A. / Lerner, C. / Mattei, P. / Neidhart, W. / Pinard, E. / Rudolph, M.G. / ...Authors: Kuhn, B. / Guba, W. / Hert, J. / Banner, D. / Bissantz, C. / Ceccarelli, S. / Haap, W. / Korner, M. / Kuglstatter, A. / Lerner, C. / Mattei, P. / Neidhart, W. / Pinard, E. / Rudolph, M.G. / Schulz-Gasch, T. / Woltering, T. / Stahl, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5f02.cif.gz | 170.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5f02.ent.gz | 135.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5f02.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5f02_validation.pdf.gz | 833.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5f02_full_validation.pdf.gz | 833.6 KB | Display | |
| Data in XML | 5f02_validation.xml.gz | 12.6 KB | Display | |
| Data in CIF | 5f02_validation.cif.gz | 18.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f0/5f02ftp://data.pdbj.org/pub/pdb/validation_reports/f0/5f02 | HTTPS FTP |
-Related structure data
| Related structure data |  5edbC  5edcC  5edeC  5edgC  5edhC  5ediC  5ezxC  5ezzC  5f00C  5f01C  5f03C  5i2rC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24191.701 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN, RESIDUES 114-333 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSL, CTSL1 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 |
| #3: Chemical | ChemComp-5T9 / (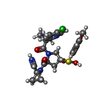  Mass: 582.499 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H29Cl2N5O5S Mass: 582.499 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H29Cl2N5O5S |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 40.78 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / Details: 0.1M MAGNESIUM FORMATE, 15 % PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Mar 13, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.43→45.78 Å / Num. obs: 34024 / % possible obs: 99.9 % / Redundancy: 6.45 % / Rsym value: 0.043 / Net I/σ(I): 12.68 |
| Reflection shell | Resolution: 1.43→1.53 Å / Redundancy: 6.16 % / Rmerge(I) obs: 0.6941 / Mean I/σ(I) obs: 1.49 / % possible all: 99.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.43→45.78 Å / Cor.coef. Fo:Fc: 0.979 / Cor.coef. Fo:Fc free: 0.965 / SU B: 3.649 / SU ML: 0.058 / Cross valid method: THROUGHOUT / ESU R: 0.076 / ESU R Free: 0.069 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.742 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.43→45.78 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|