development of primary sexual characteristics / PRC1 complex / PcG protein complex / SUMOylation of DNA methylation proteins / SUMOylation of RNA binding proteins / histone H3K9me2/3 reader activity / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / SUMOylation of DNA damage response and repair proteins / heterochromatin / SUMOylation of transcription cofactors ...development of primary sexual characteristics / PRC1 complex / PcG protein complex / SUMOylation of DNA methylation proteins / SUMOylation of RNA binding proteins / histone H3K9me2/3 reader activity / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / SUMOylation of DNA damage response and repair proteins / heterochromatin / SUMOylation of transcription cofactors / SUMOylation of chromatin organization proteins / Regulation of PTEN gene transcription / euchromatin / heterochromatin formation / Oxidative Stress Induced Senescence / cell differentiation / negative regulation of DNA-templated transcription / chromatin binding / chromatin / negative regulation of transcription by RNA polymerase II / DNA-templated transcription / DNA binding / nucleoplasm / nucleus Similarity search - Function
Chromobox protein homolog 2 / CBX family C-terminal motif / CBX family C-terminal motif / Chromo domain, conserved site / Chromo domain signature. / Chromo domain / Chromo (CHRromatin Organisation MOdifier) domain / Chromo and chromo shadow domain profile. / OB fold (Dihydrolipoamide Acetyltransferase, E2P) - #40 / Chromo/chromo shadow domain ...Chromobox protein homolog 2 / CBX family C-terminal motif / CBX family C-terminal motif / Chromo domain, conserved site / Chromo domain signature. / Chromo domain / Chromo (CHRromatin Organisation MOdifier) domain / Chromo and chromo shadow domain profile. / OB fold (Dihydrolipoamide Acetyltransferase, E2P) - #40 / Chromo/chromo shadow domain / Chromatin organization modifier domain / Chromo-like domain superfamily / OB fold (Dihydrolipoamide Acetyltransferase, E2P) / Beta Barrel / Mainly Beta Similarity search - Domain/homology
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.5418 Å / Relative weight: 1
Reflection
Resolution: 1.8→34.16 Å / Num. obs: 10262 / % possible obs: 100 % / Redundancy: 10.6 % / Rmerge(I) obs: 0.067 / Net I/σ(I): 29.2
Reflection shell
Resolution: 1.8→1.84 Å / Redundancy: 10.3 % / Rmerge(I) obs: 0.886 / Mean I/σ(I) obs: 3.1 / % possible all: 100
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0123
refinement
Aimless
0.5.1
datascaling
PDB_EXTRACT
3.2
dataextraction
PHASER
phasing
Refinement
Resolution: 1.8→34 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.945 / SU B: 3.69 / SU ML: 0.066 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.096 / ESU R Free: 0.094 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: PHENIX.ELBOW/MOGUL WAS USED TO GENERATE GEOMETRY RESTRAINTS FOR INHIBITOR BUILDING BLOCKS. JLIGAND WAS USED FOR PREPARATION OF LINK RESTRAINTS. LINK RESTRAINTS WERE MANUALLY MODIFIED, FOR ...Details: PHENIX.ELBOW/MOGUL WAS USED TO GENERATE GEOMETRY RESTRAINTS FOR INHIBITOR BUILDING BLOCKS. JLIGAND WAS USED FOR PREPARATION OF LINK RESTRAINTS. LINK RESTRAINTS WERE MANUALLY MODIFIED, FOR EXAMPLE TO ESTABLISH PLANAR GEOMETRY OF METHYL ESTER TERMINUS OF INHIBITOR. COOT WAS USED FOR INTERACTIVE MODEL BUILDING. MODEL GEOMETRY WAS EVALUATED WITH MOLPROBITY. ELECTRON DENSITY DOES NOT FULLY RESOLVE THE LIGAND'S N-EPSILON ETHYLATION AND C-TERMINAL SERINE METHYL ESTER MOIETY.
Rfactor
Num. reflection
% reflection
Rfree
0.212
506
4.9 %
Rwork
0.189
-
-
obs
0.19
9752
100 %
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION / Resolution: 1.8 Å
X-RAY DIFFRACTION / Resolution: 1.8 Å  Authors
Authors Citation
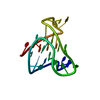
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj





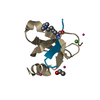
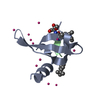
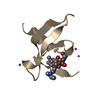
 Assembly
Assembly


 Type: Oligopeptide / Class: Inhibitor / Mass: 795.020 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.)
Type: Oligopeptide / Class: Inhibitor / Mass: 795.020 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) 
 Num. of mol.: 8 / Source method: obtained synthetically
Num. of mol.: 8 / Source method: obtained synthetically Mass: 18.015 Da / Num. of mol.: 40 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 40 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing