 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5e5r: Crystal structure of the complex between Carbonic anhydrase-like ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5e5r | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the complex between Carbonic anhydrase-like domain of PTPRG and Immunoglobulin domains 2-3 of CNTN3 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | Hydrolase/Cell Adhesion / Neural cell adhesion molecule / Receptor-type protein tyrosine phosphatase / Immunoglobulin domains / Carbonic anhydrase-like domain / Hydrolase-Cell Adhesion complex | ||||||
| Function / homology |  Function and homology information Function and homology informationPost-translational modification: synthesis of GPI-anchored proteins / transmembrane receptor protein tyrosine phosphatase activity / negative regulation of epithelial cell migration / side of membrane / protein-tyrosine-phosphatase / protein tyrosine phosphatase activity / cell surface receptor protein tyrosine kinase signaling pathway / neuron projection development / negative regulation of neuron projection development / cell adhesion ...Post-translational modification: synthesis of GPI-anchored proteins / transmembrane receptor protein tyrosine phosphatase activity / negative regulation of epithelial cell migration / side of membrane / protein-tyrosine-phosphatase / protein tyrosine phosphatase activity / cell surface receptor protein tyrosine kinase signaling pathway / neuron projection development / negative regulation of neuron projection development / cell adhesion / neuron projection / signal transduction / extracellular exosome / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.6 Å | ||||||
 Authors Authors | Nikolaienko, R.M. / Bouyain, S. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2016 Title: Structural Basis for Interactions Between Contactin Family Members and Protein-tyrosine Phosphatase Receptor Type G in Neural Tissues. Authors: Nikolaienko, R.M. / Hammel, M. / Dubreuil, V. / Zalmai, R. / Hall, D.R. / Mehzabeen, N. / Karuppan, S.J. / Harroch, S. / Stella, S.L. / Bouyain, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5e5r.cif.gz | 380.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5e5r.ent.gz | 314.7 KB | Display | PDB format |
| PDBx/mmJSON format | 5e5r.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e5/5e5rftp://data.pdbj.org/pub/pdb/validation_reports/e5/5e5r | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5e4iSC  5e4qC  5e4sC  5e52C  5e53C  5e55C  5e5uC  5e7lC  5i99C  3jxhS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 30272.697 Da / Num. of mol.: 2 / Fragment: CA domain, UNP residues 56-320 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PTPRG, PTPG / Plasmid: pET32HP / Production host:  #2: Protein | Mass: 21642.453 Da / Num. of mol.: 2 / Fragment: Immunoglobulin domains 2-3, UNP residues 124-316 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Chemical | 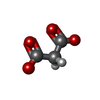  Mass: 102.046 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H2O4 Mass: 102.046 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H2O4#4: Chemical |   Mass: 46.025 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH2O2 Mass: 46.025 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH2O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 59 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 59 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.38 Å3/Da / Density % sol: 48.37 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 1% (v/v) Tacsimate pH 7.0, 20% (w/v) PEG 3350, 50mM Imidazole-HCl pH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 22-ID / Wavelength: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: May 1, 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: ROSENBAUM-ROCK DOUBLE-CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.6→50 Å / Num. obs: 31166 / % possible obs: 99.6 % / Redundancy: 11.4 % / Biso Wilson estimate: 56.08 Å2 / Rmerge(I) obs: 0.134 / Χ2: 1.02 / Net I/av σ(I): 7.237 / Net I/σ(I): 8.3 / Num. measured all: 356562 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3JXH, 5E4I Resolution: 2.6→49.149 Å / SU ML: 0.35 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 27.06 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 196.64 Å2 / Biso mean: 71.2232 Å2 / Biso min: 24.46 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.6→49.149 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 11
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
