 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5cp4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYOGENIC STRUCTURE OF P450CAM | ||||||
 Components Components | CYTOCHROME P450CAM | ||||||
 Keywords Keywords | OXIDOREDUCTASE / P450 / MONOOXYGENASE / HEME ENZYME / ELECTRON TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationcamphor 5-monooxygenase / camphor 5-monooxygenase activity / (+)-camphor catabolic process / iron ion binding / heme binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / molecular replacement / Resolution: 1.75 Å X-RAY DIFFRACTION / molecular replacement / Resolution: 1.75 Å | ||||||
 Authors Authors | Li, H. / Poulos, T.L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Understanding the role of the essential Asp251 in cytochrome p450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Authors: Vidakovic, M. / Sligar, S.G. / Li, H. / Poulos, T.L. #1: Journal: J.Mol.Biol. / Year: 1987Title: High-Resolution Crystal Structure of Cytochrome P450Cam Authors: Poulos, T.L. / Finzel, B.C. / Howard, A.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5cp4.cif.gz | 133.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5cp4.ent.gz | 103.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5cp4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cp/5cp4ftp://data.pdbj.org/pub/pdb/validation_reports/cp/5cp4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6cp4C  2cppS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 46588.879 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas putida (bacteria) / Production host: |
|---|
-Non-polymers , 5 types, 413 molecules 



| #2: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: K | ||
|---|---|---|---|
| #3: Chemical | ChemComp-HEM /  Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 | ||
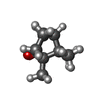
| #4: Chemical | ChemComp-CAM /  Mass: 152.233 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16O Mass: 152.233 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16O | ||
| #5: Chemical |  Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 407 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.15 Å3/Da / Density % sol: 48 % |
|---|---|
| Crystal grow | pH: 7 / Details: pH 7.0 |
| Crystal grow | *PLUS Method: other / Details: Poulos, T.L., (1996) Methods Enzymol., 272, 358. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: SIEMENS / Wavelength: 1.5418 |
| Detector | Type: SIEMENS-NICOLET X100 / Detector: AREA DETECTOR / Date: Apr 1, 1996 / Details: MIRRORS |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→30 Å / Num. obs: 31326 / % possible obs: 89.7 % / Observed criterion σ(I): 0 / Redundancy: 4 % / Rsym value: 0.058 |
| Reflection shell | Resolution: 1.75→1.9 Å / % possible all: 54.7 |
| Reflection | *PLUS Num. obs: 37112 / Num. measured all: 179587 / Rmerge(I) obs: 0.058 |
| Reflection shell | *PLUS % possible obs: 54.7 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: molecular replacement Starting model: PDB ENTRY 2CPP Resolution: 1.75→10 Å / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
