 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5btg | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of a topoisomerase II complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | Isomerase/DNA / protein-DNA complex / topoisomerase II / Isomerase-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) activity / DNA topoisomerase (ATP-hydrolysing) / DNA topological change / peptidoglycan-based cell wall / DNA-templated DNA replication / chromosome / response to antibiotic / magnesium ion binding ...DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase type II (double strand cut, ATP-hydrolyzing) activity / DNA topoisomerase (ATP-hydrolysing) / DNA topological change / peptidoglycan-based cell wall / DNA-templated DNA replication / chromosome / response to antibiotic / magnesium ion binding / ATP hydrolysis activity / DNA binding / ATP binding / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |   Mycobacterium tuberculosis (bacteria) Mycobacterium tuberculosis (bacteria)synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Blower, T.R. / Williamson, B.H. / Kerns, R.J. / Berger, J.M. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2016 Title: Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Authors: Blower, T.R. / Williamson, B.H. / Kerns, R.J. / Berger, J.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5btg.cif.gz | 945.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5btg.ent.gz | 793.4 KB | Display | PDB format |
| PDBx/mmJSON format | 5btg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 5btg_validation.pdf.gz | 1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 5btg_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 5btg_validation.xml.gz | 53.6 KB | Display | |
| Data in CIF | 5btg_validation.cif.gz | 73.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bt/5btgftp://data.pdbj.org/pub/pdb/validation_reports/bt/5btg | HTTPS FTP |
-Related structure data
| Related structure data |  5bs8SC  5btaC  5btcC  5btdC  5btfC  5btiC  5btlC  5btnC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The double-stranded DNAs are superposed copies of each other, overlaid in opposite orientations (because directionality could not be resolved). Therefore in reality, rather than in the model, the biological assembly would in fact be hexameric - four proteins and one double-stranded DNA. |
-Components
-DNA gyrase subunit ... , 2 types, 4 molecules ACBD
| #1: Protein | Mass: 56209.422 Da / Num. of mol.: 2 / Fragment: GyrA 2-500 with IGSG C-terminal tag Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) (bacteria)Strain: ATCC 25618 / H37Rv / Gene: gyrA, Rv0006, MTCY10H4.04 / Plasmid: pET-28b / Production host: #2: Protein | Mass: 28272.412 Da / Num. of mol.: 2 / Fragment: GyrB 426-675 with N-terminal SNA tag Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) (bacteria)Strain: ATCC 25618 / H37Rv / Gene: gyrB, Rv0005, MTCY10H4.03 / Plasmid: pET-28b / Production host: |
|---|
-DNA substrate 24-mer ... , 2 types, 4 molecules EHFG
| #3: DNA chain | Mass: 7417.816 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host: synthetic construct (others) #4: DNA chain | Mass: 7319.739 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host: synthetic construct (others) |
|---|
-Non-polymers , 3 types, 195 molecules 
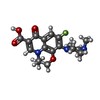

| #5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#6: Chemical |  Mass: 361.368 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H20FN3O4 Mass: 361.368 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H20FN3O4#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 189 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.76 Å3/Da / Density % sol: 55.48 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop Details: 100 mM Tris-HCl, 13-15% PEG 4000, 200 mM MgCl2, 6.25% PEG 400 PH range: 7.0 to 8.5 |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 23-ID-B / Wavelength: 1.033 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Dec 7, 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.033 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.5→46.33 Å / Num. all: 283502 / Num. obs: 283502 / % possible obs: 99.7 % / Redundancy: 3.8 % / Biso Wilson estimate: 40.67 Å2 / Rmerge F obs: 0.987 / Rmerge(I) obs: 0.1391 / Rrim(I) all: 0.162 / Χ2: 1.009 / Net I/σ(I): 8.93 / Num. measured all: 283517 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5BS8 Resolution: 2.5→46.325 Å / SU ML: 0.37 / Cross valid method: FREE R-VALUE / σ(F): 1 / Phase error: 27.67 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 255.2 Å2 / Biso mean: 65.8645 Å2 / Biso min: 14 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.5→46.325 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 30
|
