 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5agu: The sliding clamp of Mycobacterium tuberculosis in complex with a... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5agu | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | The sliding clamp of Mycobacterium tuberculosis in complex with a natural product. | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSFERASE / DNAN / DNA POLYMERASE / MYCOBACTERIUM TUBERCULOSIS / TUBERCULOSIS / NATURAL PRODUCT / SLIDING CLAMP | |||||||||
| Function / homology |  Function and homology information Function and homology informationDNA polymerase III complex / DNA strand elongation involved in DNA replication / 3'-5' exonuclease activity / peptidoglycan-based cell wall / DNA-directed DNA polymerase activity / response to antibiotic / DNA binding / extracellular region / identical protein binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  MYCOBACTERIUM TUBERCULOSIS H37RV (bacteria)STREPTOMYCES CAELICUS (bacteria) MYCOBACTERIUM TUBERCULOSIS H37RV (bacteria)STREPTOMYCES CAELICUS (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.173 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.173 Å | |||||||||
 Authors Authors | Lukat, P. / Kling, A. / Heinz, D.W. / Mueller, R. | |||||||||
 Citation Citation | Journal: Science / Year: 2015 Title: Antibiotics. Targeting Dnan for Tuberculosis Therapy Using Novel Griselimycins. Authors: Kling, A. / Lukat, P. / Almeida, D.V. / Bauer, A. / Fontaine, E. / Sordello, S. / Zaburannyi, N. / Herrmann, J. / Wenzel, S.C. / Konig, C. / Ammerman, N.C. / Barrio, M.B. / Borchers, K. / ...Authors: Kling, A. / Lukat, P. / Almeida, D.V. / Bauer, A. / Fontaine, E. / Sordello, S. / Zaburannyi, N. / Herrmann, J. / Wenzel, S.C. / Konig, C. / Ammerman, N.C. / Barrio, M.B. / Borchers, K. / Bordon-Pallier, F. / Bronstrup, M. / Courtemanche, G. / Gerlitz, M. / Geslin, M. / Hammann, P. / Heinz, D.W. / Hoffmann, H. / Klieber, S. / Kohlmann, M. / Kurz, M. / Lair, C. / Matter, H. / Nuermberger, E. / Tyagi, S. / Fraisse, L. / Grosset, J.H. / Lagrange, S. / Muller, R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5agu.cif.gz | 305.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5agu.ent.gz | 249.1 KB | Display | PDB format |
| PDBx/mmJSON format | 5agu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ag/5aguftp://data.pdbj.org/pub/pdb/validation_reports/ag/5agu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5agvC  5ah2C  5ah4C  3p16S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.99814, -0.0013, 0.06094), Vector: |
-Components
| #1: Protein | Mass: 42479.207 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) MYCOBACTERIUM TUBERCULOSIS H37RV (bacteria)Plasmid: PVP008_DNANMTB / Production host: References: UniProt: I6XU56, UniProt: P9WNU1*PLUS, DNA-directed DNA polymerase #2: Protein/peptide |   Type: Peptide-like / Class: Inhibitor / Mass: 1115.449 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) STREPTOMYCES CAELICUS (bacteria) / References: ACE-MVA-MP8-NZC-LEU-MP8-LEU-MVA-PRO-MLU-GLY Type: Peptide-like / Class: Inhibitor / Mass: 1115.449 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) STREPTOMYCES CAELICUS (bacteria) / References: ACE-MVA-MP8-NZC-LEU-MP8-LEU-MVA-PRO-MLU-GLY#3: Chemical | ChemComp-BU3 / ( | 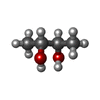  Mass: 90.121 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2 Mass: 90.121 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2#4: Chemical | ChemComp-TRS / |   Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 357 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 357 / Source method: isolated from a natural source / Formula: H2OSequence details | FIRST 4 N-TERMINAL RESIDUES ARE REMAINING FROM THE TEV- CLEAVAGE SITE AFTER PROTEOLYSI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 49 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: 21.2 % (W/V) PEG 3000, 100 MM TRIS-HCL PH 7.67. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.918409 / Beamline: 14.1 / Wavelength: 0.918409 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Sep 24, 2014 / Details: MIRRORS |
| Radiation | Monochromator: KMC-1 / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.918409 Å / Relative weight: 1 |
| Reflection | Resolution: 2.17→199.3 Å / Num. obs: 45449 / % possible obs: 100 % / Observed criterion σ(I): -3 / Redundancy: 20.8 % / Biso Wilson estimate: 32.62 Å2 / Rmerge(I) obs: 0.2 / Net I/σ(I): 13.2 |
| Reflection shell | Resolution: 2.17→2.18 Å / Redundancy: 20.7 % / Rmerge(I) obs: 1.5 / Mean I/σ(I) obs: 2.1 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3P16 Resolution: 2.173→70.841 Å / SU ML: 0.26 / σ(F): 1.35 / Phase error: 23.99 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.24 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.173→70.841 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
