Hydrolases; Glycosylases; Glycosidases, i.e. enzymes that hydrolyse O- and S-glycosyl compounds / polysaccharide catabolic process / hydrolase activity, hydrolyzing O-glycosyl compounds Similarity search - Function
Glycoside hydrolase, family 8, conserved site / Glycosyl hydrolases family 8 signature. / NedA-like, galactose-binding domain / Glycoside hydrolase, family 8 / Glycosyl hydrolases family 8 / Coagulation factor 5/8 C-terminal domain, discoidin domain / Coagulation factors 5/8 type C domain (FA58C) profile. / F5/8 type C domain / Coagulation factor 5/8 C-terminal domain / Six-hairpin glycosidase-like superfamily ...Glycoside hydrolase, family 8, conserved site / Glycosyl hydrolases family 8 signature. / NedA-like, galactose-binding domain / Glycoside hydrolase, family 8 / Glycosyl hydrolases family 8 / Coagulation factor 5/8 C-terminal domain, discoidin domain / Coagulation factors 5/8 type C domain (FA58C) profile. / F5/8 type C domain / Coagulation factor 5/8 C-terminal domain / Six-hairpin glycosidase-like superfamily / Six-hairpin glycosidase superfamily / Galactose-binding domain-like / Fibronectin type III domain / Fibronectin type 3 domain / Fibronectin type-III domain profile. / Galactose-binding-like domain superfamily / Fibronectin type III / Fibronectin type III superfamily / Jelly Rolls / Immunoglobulin-like fold / Sandwich / Mainly Beta Similarity search - Domain/homology
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Paenibacillus fukuinensis (bacteria)
Paenibacillus fukuinensis (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Japan, 1items
Japan, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj




 Assembly
Assembly

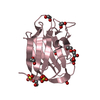

 Mass: 62.068 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 18 / Source method: obtained synthetically / Formula: C2H6O2
 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 625 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 625 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing