 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
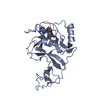
Yorodumi- PDB-4ynm: ASH1L wild-type SET domain in complex with S-adenosyl methionine (SAM) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ynm | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | ASH1L wild-type SET domain in complex with S-adenosyl methionine (SAM) | ||||||||||||||||||||||||
 Components Components | Histone-lysine N-methyltransferase ASH1L | ||||||||||||||||||||||||
 Keywords Keywords | TRANSFERASE / histone methylation / SET domain | ||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationuterine gland development / tarsal gland development / histone H3K9 monomethyltransferase activity / [histone H3]-lysine36 N-trimethyltransferase / histone H3K36 trimethyltransferase activity / [histone H3]-lysine9 N-methyltransferase / uterus morphogenesis / histone H3K9 methyltransferase activity / histone H3K9me2 methyltransferase activity / flagellated sperm motility ...uterine gland development / tarsal gland development / histone H3K9 monomethyltransferase activity / [histone H3]-lysine36 N-trimethyltransferase / histone H3K36 trimethyltransferase activity / [histone H3]-lysine9 N-methyltransferase / uterus morphogenesis / histone H3K9 methyltransferase activity / histone H3K9me2 methyltransferase activity / flagellated sperm motility / histone H3K36 methyltransferase activity / histone H3K4 methyltransferase activity / histone H3 methyltransferase activity / negative regulation of acute inflammatory response / decidualization / bicellular tight junction / single fertilization / negative regulation of MAPK cascade / post-embryonic development / skeletal system development / PKMTs methylate histone lysines / MAPK cascade / chromosome / methylation / transcription by RNA polymerase II / inflammatory response / chromatin binding / regulation of DNA-templated transcription / Golgi apparatus / positive regulation of transcription by RNA polymerase II / DNA binding / nucleoplasm / nucleus / metal ion binding Similarity search - Function | ||||||||||||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||||||||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.19 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.19 Å | ||||||||||||||||||||||||
 Authors Authors | Rogawski, D.S. / Ndoj, J. / Cho, H.-J. / Maillard, I. / Grembecka, J. / Cierpicki, T. | ||||||||||||||||||||||||
| Funding support |  United States, 7items United States, 7items
| ||||||||||||||||||||||||
 Citation Citation | Journal: Biochemistry / Year: 2015 Title: Two Loops Undergoing Concerted Dynamics Regulate the Activity of the ASH1L Histone Methyltransferase. Authors: Rogawski, D.S. / Ndoj, J. / Cho, H.J. / Maillard, I. / Grembecka, J. / Cierpicki, T. | ||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ynm.cif.gz | 103.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ynm.ent.gz | 77.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4ynm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4ynm_validation.pdf.gz | 1.1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4ynm_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 4ynm_validation.xml.gz | 20.2 KB | Display | |
| Data in CIF | 4ynm_validation.cif.gz | 27 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yn/4ynmftp://data.pdbj.org/pub/pdb/validation_reports/yn/4ynm | HTTPS FTP |
-Related structure data
| Related structure data |  4ynpC  4ypaC  4ypeC  4ypuC  3opeS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS ensembles :
|
-Components
| #1: Protein | Mass: 26016.516 Da / Num. of mol.: 2 / Fragment: SET domain (UNP residues 2074-2293) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: ASH1L, KIAA1420, KMT2H / Production host:  References: UniProt: Q9NR48, histone-lysine N-methyltransferase #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Zn#3: Chemical | 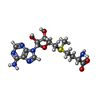  Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 104 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 104 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.41 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / Details: PEG 3350, 20 mM Tris / PH range: 7.3-7.8 |
-Data collection
| Diffraction | Mean temperature: 123 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 21-ID-D / Wavelength: 0.97872 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Dec 8, 2012 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97872 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.19→50 Å / Num. obs: 25099 / % possible obs: 99.9 % / Redundancy: 9.1 % / Rmerge(I) obs: 0.095 / Rpim(I) all: 0.033 / Rrim(I) all: 0.101 / Χ2: 1.645 / Net I/av σ(I): 33.696 / Net I/σ(I): 7.8 / Num. measured all: 228388 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3OPE Resolution: 2.19→46.82 Å / Cor.coef. Fo:Fc: 0.926 / Cor.coef. Fo:Fc free: 0.913 / SU B: 6.875 / SU ML: 0.178 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.323 / ESU R Free: 0.238 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 95.31 Å2 / Biso mean: 42.453 Å2 / Biso min: 20.1 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.19→46.82 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.192→2.249 Å / Total num. of bins used: 20
|
