Entry Database : PDB / ID : 4xojTitle Structure of bovine trypsin in complex with analogues of sunflower inhibitor 1 (SFTI-1) Cationic trypsin Trypsin inhibitor 1 Keywords / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Bos taurus (domestic cattle)Helianthus annuus (common sunflower)Method / / / Resolution : 0.91 Å Authors Golik, P. / Malicki, S. / Grudnik, P. / Karna, N. / Debowski, D. / Legowska, A. / Wladyka, B. / Gitlin, A. / Brzozowski, K. / Dubin, G. / Rolka, K. Journal : Chembiochem / Year : 2015Title : Investigation of Serine-Proteinase-Catalyzed Peptide Splicing in Analogues of Sunflower Trypsin Inhibitor 1 (SFTI-1).Authors : Karna, N. / Legowska, A. / Malicki, S. / Debowski, D. / Golik, P. / Gitlin, A. / Grudnik, P. / Wladyka, B. / Brzozowski, K. / Dubin, G. / Rolka, K. History Deposition Jan 16, 2015 Deposition site / Processing site Revision 1.0 Aug 12, 2015 Provider / Type Revision 1.1 Sep 23, 2015 Group Revision 1.2 Aug 9, 2017 Group / Database references / Category / diffrn_sourceItem / _diffrn_source.pdbx_synchrotron_siteRevision 1.3 Jan 10, 2024 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_alt_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_alt_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_ptnr2_label_alt_id / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id Revision 1.4 Oct 9, 2024 Group / Category / pdbx_modification_feature
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information

 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj


 Assembly
Assembly





 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 22.990 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Na
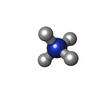
Mass: 22.990 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Na Mass: 18.038 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H4N
Mass: 18.038 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: H4N Sample preparation
Sample preparation / Beamline: P13 (MX1) / Wavelength: 1 Å
/ Beamline: P13 (MX1) / Wavelength: 1 Å Processing
Processing