Entry Database : PDB / ID : 4tsrTitle The Complex Structure of Mutant Phytase with IHS Periplasmic AppA protein Keywords / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / Biological species Escherichia coli (E. coli)Method / / / Resolution : 2.07 Å Authors Wu, T.H. / Chen, C.C. / Huang, C.H. / Guo, R.T. Journal : To Be Published Title : The Complex Structure of mutant Phytase with IHSAuthors : Wu, T.H. / Chen, C.C. / Huang, C.H. / Guo, R.T. History Deposition Jun 19, 2014 Deposition site / Processing site Revision 1.0 May 6, 2015 Provider / Type Revision 1.1 Sep 27, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description / Source and taxonomy / Structure summary Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / citation / database_2 / entity_src_gen / pdbx_database_status / pdbx_initial_refinement_model / pdbx_struct_conn_angle / pdbx_struct_oper_list / refine_hist / struct_conn / struct_keywords Item _citation.journal_id_CSD / _database_2.pdbx_DOI ... _citation.journal_id_CSD / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _entity_src_gen.pdbx_alt_source_flag / _pdbx_database_status.pdb_format_compatible / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.value / _pdbx_struct_oper_list.symmetry_operation / _refine_hist.number_atoms_solvent / _refine_hist.pdbx_number_atoms_ligand / _refine_hist.pdbx_number_atoms_nucleic_acid / _refine_hist.pdbx_number_atoms_protein / _struct_conn.pdbx_dist_value / _struct_conn.ptnr2_auth_seq_id / _struct_keywords.text Revision 1.2 Oct 23, 2024 Group / Category / pdbx_modification_feature
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj


 Assembly
Assembly
 Komagataella pastoris CBS 7435 (fungus) / References: UniProt: P07102, acid phosphatase, 4-phytase
Komagataella pastoris CBS 7435 (fungus) / References: UniProt: P07102, acid phosphatase, 4-phytase
 Mass: 58.693 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ni
Mass: 58.693 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ni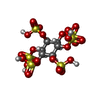
 Mass: 660.535 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H12O24S6
Mass: 660.535 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H12O24S6 Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL13C1 / Wavelength: 0.97622 Å
/ Beamline: BL13C1 / Wavelength: 0.97622 Å Processing
Processing