| 登録情報 | データベース: PDB / ID: 4rle
|
|---|
| タイトル | Crystal structure of the c-di-AMP binding PII-like protein DarA |
|---|
 要素 要素 | Uncharacterized protein YaaQ |
|---|
 キーワード キーワード | UNKNOWN FUNCTION / PII-like / cdiAMP |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
Cyclic-di-AMP receptor / Cyclic-di-AMP receptor / Alpha-Beta Plaits - #120 / Nitrogen regulatory PII-like, alpha/beta / Nitrogen regulatory protein PII/ATP phosphoribosyltransferase, C-terminal / Alpha-Beta Plaits / 2-Layer Sandwich / Alpha Beta類似検索 - ドメイン・相同性 Chem-2BA / NICKEL (II) ION / Cyclic di-AMP receptor A類似検索 - 構成要素 |
|---|
| 生物種 |   Bacillus subtilis (枯草菌) Bacillus subtilis (枯草菌) |
|---|
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.3 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.3 Å |
|---|
 データ登録者 データ登録者 | Dickmanns, A. / Neumann, P. / Ficner, R. |
|---|
 引用 引用 | ジャーナル: J.Biol.Chem. / 年: 2015
タイトル: Identification, Characterization, and Structure Analysis of the Cyclic di-AMP-binding PII-like Signal Transduction Protein DarA.
著者: Gundlach, J. / Dickmanns, A. / Schroder-Tittmann, K. / Neumann, P. / Kaesler, J. / Kampf, J. / Herzberg, C. / Hammer, E. / Schwede, F. / Kaever, V. / Tittmann, K. / Stulke, J. / Ficner, R. |
|---|
| 履歴 | | 登録 | 2014年10月16日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2014年12月3日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2014年12月17日 | Group: Database references |
|---|
| 改定 1.2 | 2015年2月11日 | Group: Database references |
|---|
| 改定 1.3 | 2019年9月4日 | Group: Data collection / カテゴリ: reflns / Item: _reflns.pdbx_Rsym_value |
|---|
| 改定 1.4 | 2023年9月20日 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Refinement description / Structure summary
カテゴリ: chem_comp / chem_comp_atom ...chem_comp / chem_comp_atom / chem_comp_bond / database_2 / entity / pdbx_entity_nonpoly / pdbx_initial_refinement_model / struct_ref_seq_dif / struct_site
Item: _chem_comp.name / _database_2.pdbx_DOI ..._chem_comp.name / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _entity.pdbx_description / _pdbx_entity_nonpoly.name / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード








 PDBj
PDBj 集合体
集合体

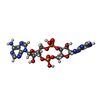
 分子量: 658.412 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C20H24N10O12P2
分子量: 658.412 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C20H24N10O12P2
 分子量: 58.693 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ni
分子量: 58.693 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ni 分子量: 18.015 Da / 分子数: 145 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 145 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: ID23-1 / 波長: 0.91 Å
/ ビームライン: ID23-1 / 波長: 0.91 Å 解析
解析