Formation of the active cofactor, UDP-glucuronate / UTP-glucose-1-phosphate uridylyltransferase / UTP:glucose-1-phosphate uridylyltransferase activity / UDP-alpha-D-glucose metabolic process / glycogen biosynthetic process / glycogen metabolic process / Glycogen synthesis / brain development / extracellular exosome / metal ion binding ...Formation of the active cofactor, UDP-glucuronate / UTP-glucose-1-phosphate uridylyltransferase / UTP:glucose-1-phosphate uridylyltransferase activity / UDP-alpha-D-glucose metabolic process / glycogen biosynthetic process / glycogen metabolic process / Glycogen synthesis / brain development / extracellular exosome / metal ion binding / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function
UTP--glucose-1-phosphate uridylyltransferase / UDPGP family / UTP--glucose-1-phosphate uridylyltransferase / Hexapeptide repeat proteins / UDP N-Acetylglucosamine Acyltransferase; domain 1 / 3 Solenoid / Spore Coat Polysaccharide Biosynthesis Protein SpsA; Chain A / Spore Coat Polysaccharide Biosynthesis Protein SpsA; Chain A / Nucleotide-diphospho-sugar transferases / Alpha-Beta Complex ...UTP--glucose-1-phosphate uridylyltransferase / UDPGP family / UTP--glucose-1-phosphate uridylyltransferase / Hexapeptide repeat proteins / UDP N-Acetylglucosamine Acyltransferase; domain 1 / 3 Solenoid / Spore Coat Polysaccharide Biosynthesis Protein SpsA; Chain A / Spore Coat Polysaccharide Biosynthesis Protein SpsA; Chain A / Nucleotide-diphospho-sugar transferases / Alpha-Beta Complex / Mainly Beta / Alpha Beta Similarity search - Domain/homology
Two cyclic C4 tetramers are stacked onto each other and are related by twofold symmetry axes creating the octameric structure with dihedral symmetry D4
Mass: 18.015 Da / Num. of mol.: 117 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.68 Å3/Da / Density % sol: 66.62 %
Crystal grow
Temperature: 281.15 K / pH: 7.5 Details: The protein sample contained 10.3 mg/ml of hUGP, 50 mM HEPES pH 7.5, 5 mM MgCl2, 1 mM EDTA, 20% (w/v) sucrose and 4 mM UDP-Glc. 1 l of the hUGP UDP-Glc complex was mixed 1:1 with the ...Details: The protein sample contained 10.3 mg/ml of hUGP, 50 mM HEPES pH 7.5, 5 mM MgCl2, 1 mM EDTA, 20% (w/v) sucrose and 4 mM UDP-Glc. 1 l of the hUGP UDP-Glc complex was mixed 1:1 with the reservoir solution containing 100 mM sodium acetate buffer pH 4.8, 520 mM zinc acetate, 6% (w/v) aminocaproic acid and 75 mM ammonium sulfate. , VAPOR DIFFUSION, SITTING DROP, temperature 281.15K
Monochromator: DIAMOND (111), GE(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9334 Å / Relative weight: 1
Reflection
Resolution: 3.3→47.623 Å / Num. obs: 58213 / % possible obs: 99.7 % / Observed criterion σ(I): 2 / Redundancy: 12.3 % / Biso Wilson estimate: 96.27 Å2
-
Processing
Software
Name
Version
Classification
MxCuBE
datacollection
AMoRE
phasing
PHENIX
(phenix.refine: 1.9_1692)
refinement
XDS
datareduction
SADABS
datascaling
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 3.35→47.62 Å / SU ML: 0.41 / σ(F): 1.34 / Phase error: 24.49 / Stereochemistry target values: ML Details: Author stated the following: In the crystals of hUGP1-UDP-Glc complex the electron density for the substrate (UPG) as well as for some other parts of the structure was observed to be rather ...Details: Author stated the following: In the crystals of hUGP1-UDP-Glc complex the electron density for the substrate (UPG) as well as for some other parts of the structure was observed to be rather weak. It may in part be explained by a certain degree of disorder within the unit cell, which is reflected in a high value of the Wilson B-factor (108.6 A**2) and a weaker electron density for the protein chain D. However, the partial electron density observed for the UPG ribose and the glucose moieties indicated that the substrate is present in the active site of protein chain A. To improve the quality of the electron density, especially in its weak regions, we performed a phase improvement procedure using the density modification (DM) methods in the following way. The DM was performed in a solvent flattening mode using Hendrickson-Lattman coefficients from the refined protein model with omitted substrate and ligands as an initial approximation. After several cycles of the DM, we observed a noticeable increase of the figure-of-merit (15% in a high-resolution shell). The resulting weighted structure factor and modified phases from DM procedure were used to calculate the composite 2Fo-Fc electron density which you can see on the figure attached to this message. The DM-improved ligand-omit map allowed to determine the binding mode of UPG unambiguously. In addition to the UPG, the DM map allowed to resolve several other weak density regions which were not clearly interpretable before. The described method of improvement of the weak electron density regions has been successfully applied to resolve the disordered protein regions in a publication: Fedorov R, Witte G, Urbanke C, Manstein DJ, Curth U. 3D structure of Thermus aquaticus single-stranded DNA-binding protein gives insight into the functioning of SSB proteins. (2006) Nucleic Acids Res 34, 6708-6717. and in a number of application notes of our group together with Bruker company. The considerable increase of the electron density quality and the value of figure-of-merit in the particular case of hUGP1.UDP-Glc complex structure can be explained by a high solvent content in the hUGP1.UDP-Glc complex crystals (Vs = 67.7%), which makes the phase improvement by solvent flattening procedure very efficient.
Rfactor
Num. reflection
% reflection
Rfree
0.247
2580
5.06 %
Rwork
0.199
-
-
obs
0.202
50946
99.9 %
all
-
49306
-
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
Refinement step
Cycle: LAST / Resolution: 3.35→47.62 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
15292
0
182
117
15591
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.008
15718
X-RAY DIFFRACTION
f_angle_d
1.101
21248
X-RAY DIFFRACTION
f_dihedral_angle_d
16.348
5912
X-RAY DIFFRACTION
f_chiral_restr
0.042
2428
X-RAY DIFFRACTION
f_plane_restr
0.006
2714
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
3.35-3.4144
0.3415
140
0.2804
2637
X-RAY DIFFRACTION
100
3.4144-3.4841
0.3087
135
0.2671
2666
X-RAY DIFFRACTION
100
3.4841-3.5598
0.2991
159
0.2299
2643
X-RAY DIFFRACTION
100
3.5598-3.6426
0.2906
141
0.2179
2656
X-RAY DIFFRACTION
100
3.6426-3.7337
0.2656
159
0.2135
2612
X-RAY DIFFRACTION
100
3.7337-3.8346
0.2284
124
0.2076
2672
X-RAY DIFFRACTION
100
3.8346-3.9474
0.2541
130
0.1848
2683
X-RAY DIFFRACTION
100
3.9474-4.0747
0.2799
136
0.1706
2660
X-RAY DIFFRACTION
100
4.0747-4.2202
0.2067
163
0.1706
2652
X-RAY DIFFRACTION
100
4.2202-4.3891
0.226
161
0.1486
2630
X-RAY DIFFRACTION
100
4.3891-4.5887
0.2181
140
0.15
2689
X-RAY DIFFRACTION
100
4.5887-4.8304
0.2056
138
0.1524
2676
X-RAY DIFFRACTION
100
4.8304-5.1327
0.2069
145
0.1542
2699
X-RAY DIFFRACTION
100
5.1327-5.5285
0.232
148
0.1706
2687
X-RAY DIFFRACTION
100
5.5285-6.0838
0.2253
125
0.1903
2726
X-RAY DIFFRACTION
100
6.0838-6.9618
0.3648
136
0.243
2751
X-RAY DIFFRACTION
100
6.9618-8.7622
0.249
167
0.2304
2740
X-RAY DIFFRACTION
100
8.7622-47.6279
0.233
133
0.2295
2887
X-RAY DIFFRACTION
99
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
1.4336
0.6327
-0.5328
2.4501
-0.2041
2.2142
0.2606
0.1859
0.2441
-0.1454
-0.1926
-0.317
-0.2938
0.4286
0.0385
1.3017
0.6193
0.1773
1.0095
0.1505
0.4089
29.9816
-23.0143
31.0992
2
1.1396
-0.3653
-0.2837
1.7929
0.5103
1.7256
0.1361
0.4033
0.0333
-0.6717
-0.2727
-0.107
-0.0632
-0.1429
0.1979
1.5382
0.6163
0.238
1.0113
0.1903
0.6275
63.2993
-93.948
27.7658
3
2.303
-0.5124
0.0229
1.6239
-0.0827
4.1815
0.1014
0.7922
-0.0165
-0.3951
-0.1194
0.3102
-0.3792
-1.0061
0.0942
1.1944
0.4488
-0.014
1.1929
-0.1277
0.685
14.6736
-76.74
11.318
4
4.2572
-0.4473
-1.0949
1.8734
-0.3627
2.9119
0.0437
0.3199
0.5942
-0.4008
-0.2299
-0.9655
-0.1878
1.1757
0.1845
1.3752
0.1934
0.2515
1.1418
0.312
1.1083
78.5088
-40.1776
47.6058
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
1
X-RAY DIFFRACTION
1
(chain 'A' andresid24through508)
2
X-RAY DIFFRACTION
2
(chain 'B' andresid24through508)
3
X-RAY DIFFRACTION
3
(chain 'C' andresid24through508)
4
X-RAY DIFFRACTION
4
(chain 'D' andresid24through508)
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj



 Assembly
Assembly


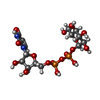





 Mass: 566.302 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H24N2O17P2
Mass: 566.302 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H24N2O17P2 Mass: 96.063 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: SO4 Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2
Mass: 59.044 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 62.068 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 65.409 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: Zn Sample preparation
Sample preparation / Beamline: ID14-1 / Wavelength: 0.9334
/ Beamline: ID14-1 / Wavelength: 0.9334  Processing
Processing