bBAF complex / npBAF complex / brahma complex / nBAF complex / Formation of the canonical BAF (cBAF) complex / Formation of neuronal progenitor and neuronal BAF (npBAF and nBAF) / Formation of the polybromo-BAF (pBAF) complex / Formation of the non-canonical BAF (ncBAF) complex / GBAF complex / regulation of G0 to G1 transition ...bBAF complex / npBAF complex / brahma complex / nBAF complex / Formation of the canonical BAF (cBAF) complex / Formation of neuronal progenitor and neuronal BAF (npBAF and nBAF) / Formation of the polybromo-BAF (pBAF) complex / Formation of the non-canonical BAF (ncBAF) complex / GBAF complex / regulation of G0 to G1 transition / nucleosome array spacer activity / intermediate filament cytoskeleton / RSC-type complex / regulation of nucleotide-excision repair / SWI/SNF complex / regulation of mitotic metaphase/anaphase transition / positive regulation of T cell differentiation / positive regulation of double-strand break repair / positive regulation of stem cell population maintenance / Regulation of MITF-M-dependent genes involved in pigmentation / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / regulation of G1/S transition of mitotic cell cycle / spermatid development / negative regulation of cell differentiation / positive regulation of myoblast differentiation / ATP-dependent activity, acting on DNA / helicase activity / positive regulation of cell differentiation / Regulation of endogenous retroelements by Piwi-interacting RNAs (piRNAs) / negative regulation of cell growth / Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to facilitate cellular and subcellular movement / RMTs methylate histone arginines / nervous system development / heterochromatin formation / histone binding / transcription coactivator activity / transcription cis-regulatory region binding / chromatin remodeling / negative regulation of cell population proliferation / negative regulation of DNA-templated transcription / positive regulation of cell population proliferation / chromatin binding / regulation of transcription by RNA polymerase II / regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / chromatin / negative regulation of transcription by RNA polymerase II / ATP hydrolysis activity / positive regulation of transcription by RNA polymerase II / DNA binding / nucleoplasm / ATP binding / nucleus Similarity search - Function
SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2 / cDNA FLJ36757 fis, clone UTERU2018522, highly similar to Human transcriptional activator hSNF2a Similarity search - Component
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
















 PDBj
PDBj

 Assembly
Assembly
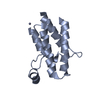




 Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I03 / Wavelength: 0.9763 Å
/ Beamline: I03 / Wavelength: 0.9763 Å Processing
Processing