error-prone translesion synthesis / DNA-templated DNA replication / DNA-directed DNA polymerase / damaged DNA binding / DNA-directed DNA polymerase activity / DNA repair / magnesium ion binding / cytoplasm Similarity search - Function
DNA polymerase type-Y, HhH motif / IMS family HHH motif / DNA polymerase IV / : / DNA polymerase, Y-family, little finger domain / MutS, DNA mismatch repair protein, domain I - #60 / MutS, DNA mismatch repair protein, domain I / DNA polymerase, Y-family, little finger domain / impB/mucB/samB family C-terminal domain / UmuC domain ...DNA polymerase type-Y, HhH motif / IMS family HHH motif / DNA polymerase IV / : / DNA polymerase, Y-family, little finger domain / MutS, DNA mismatch repair protein, domain I - #60 / MutS, DNA mismatch repair protein, domain I / DNA polymerase, Y-family, little finger domain / impB/mucB/samB family C-terminal domain / UmuC domain / DNA polymerase, Y-family, little finger domain superfamily / impB/mucB/samB family / UmuC domain profile. / Reverse transcriptase/Diguanylate cyclase domain / Dna Ligase; domain 1 / 5' to 3' exonuclease, C-terminal subdomain / DNA polymerase; domain 1 / Reverse transcriptase/Diguanylate cyclase domain / Alpha-Beta Plaits / DNA/RNA polymerase superfamily / 2-Layer Sandwich / Orthogonal Bundle / 3-Layer(aba) Sandwich / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
A: DNA polymerase IV C: DNA (5'-D(P*GP*GP*CP*TP*AP*CP*AP*GP*GP*AP*CP*TP*C)-3') D: DNA (5'-D(P*CP*AP*GP*GP*AP*GP*TP*CP*CP*TP*GP*TP*AP*GP*CP*C)-3') hetero molecules
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Sulfolobus solfataricus (archaea)
Sulfolobus solfataricus (archaea) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

























 PDBj
PDBj






































 Assembly
Assembly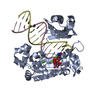

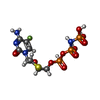


 Mass: 486.202 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H14FN4O11P3S
Mass: 486.202 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H14FN4O11P3S Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca Sample preparation
Sample preparation / Beamline: 31-ID / Wavelength: 0.97931
/ Beamline: 31-ID / Wavelength: 0.97931  Processing
Processing