 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4p3w: Crystal structure of the human filamin A Ig-like domains 20-21 in... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4p3w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human filamin A Ig-like domains 20-21 in complex with migfilin peptide | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CELL ADHESION / cytoskeleton / adhesion / immunoglobulin-like / actin binding protein | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of integrin activation / regulation of membrane repolarization during atrial cardiac muscle cell action potential / regulation of membrane repolarization during cardiac muscle cell action potential / establishment of Sertoli cell barrier / formation of radial glial scaffolds / Myb complex / filamin binding / adenylate cyclase-inhibiting dopamine receptor signaling pathway / positive regulation of integrin-mediated signaling pathway / blood coagulation, intrinsic pathway ...regulation of integrin activation / regulation of membrane repolarization during atrial cardiac muscle cell action potential / regulation of membrane repolarization during cardiac muscle cell action potential / establishment of Sertoli cell barrier / formation of radial glial scaffolds / Myb complex / filamin binding / adenylate cyclase-inhibiting dopamine receptor signaling pathway / positive regulation of integrin-mediated signaling pathway / blood coagulation, intrinsic pathway / OAS antiviral response / protein localization to bicellular tight junction / actin crosslink formation / positive regulation of actin filament bundle assembly / positive regulation of neuron migration / tubulin deacetylation / megakaryocyte development / Cell-extracellular matrix interactions / positive regulation of platelet activation / positive regulation of potassium ion transmembrane transport / apical dendrite / Fc-gamma receptor I complex binding / positive regulation of neural precursor cell proliferation / protein localization to cell surface / negative regulation of transcription by RNA polymerase I / podosome / wound healing, spreading of cells / GP1b-IX-V activation signalling / SMAD binding / receptor clustering / cortical cytoskeleton / RHO GTPases activate PAKs / semaphorin-plexin signaling pathway / mitotic spindle assembly / cilium assembly / potassium channel regulator activity / release of sequestered calcium ion into cytosol / positive regulation of substrate adhesion-dependent cell spreading / stress fiber / regulation of cell migration / protein localization to plasma membrane / cell periphery / dendritic shaft / actin filament / establishment of protein localization / cell-cell adhesion / negative regulation of protein catabolic process / protein sequestering activity / cerebral cortex development / positive regulation of protein import into nucleus / cell junction / platelet aggregation / mRNA transcription by RNA polymerase II / G protein-coupled receptor binding / small GTPase binding / kinase binding / Z disc / cell-cell junction / actin filament binding / actin cytoskeleton / Platelet degranulation / regulation of cell shape / growth cone / actin cytoskeleton organization / GTPase binding / DNA-binding transcription factor binding / perikaryon / transmembrane transporter binding / positive regulation of canonical NF-kappaB signal transduction / postsynapse / protein stabilization / cadherin binding / focal adhesion / nucleolus / negative regulation of apoptotic process / perinuclear region of cytoplasm / glutamatergic synapse / protein homodimerization activity / RNA binding / extracellular exosome / extracellular region / membrane / metal ion binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Seppala, J. / Pentikainen, U. / Ylanne, J. | ||||||
 Citation Citation | Journal: To Be Published Title: Crystal structure of the human filamin A Ig-like domains 20-21 in complex with migfilin peptide Authors: Seppala, J. / Pentikainen, U. / Ylanne, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4p3w.cif.gz | 411.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4p3w.ent.gz | 336.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4p3w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p3/4p3wftp://data.pdbj.org/pub/pdb/validation_reports/p3/4p3w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2brqS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| 2 | 
| |||||||||||||||
| 3 | 
| |||||||||||||||
| 4 | 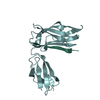
| |||||||||||||||
| 5 | 
| |||||||||||||||
| 6 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein / Protein/peptide , 2 types, 12 molecules ABEFDCGHKLJI
| #1: Protein | Mass: 19023.158 Da / Num. of mol.: 6 / Fragment: Filamin repeats 20 and 21, residues 2152-2329 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: FLNA, FLN, FLN1 / Plasmid: pGEX4T3-TEV / Production host:  #2: Protein/peptide | Mass: 2613.020 Da / Num. of mol.: 6 / Fragment: residues 5-28 / Source method: obtained synthetically / Source: (synth.) Homo sapiens (human) / References: UniProt: Q8WUP2 |
|---|
-Non-polymers , 4 types, 127 molecules 



| #3: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-PR /  Mass: 140.908 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Pr Mass: 140.908 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Pr#5: Chemical |  Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 117 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47.09 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 8 / Details: 0.1 M MES pH 6, 1.9 M (NH4)2SO4, 0.1 M (CH3CO2)3Pr |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.9334 Å / Beamline: ID14-1 / Wavelength: 0.9334 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jul 13, 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Ge(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9334 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.8→45.36 Å / Num. obs: 103427 / % possible obs: 97.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 3.426 % / Biso Wilson estimate: 40.614 Å2 / Rmerge F obs: 0.999 / Rmerge(I) obs: 0.053 / Rrim(I) all: 0.062 / Rsym value: 0.062 / Χ2: 0.985 / Net I/σ(I): 14.07 / Num. measured all: 354375 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2BRQ Resolution: 2→43.86 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.954 / WRfactor Rfree: 0.2103 / WRfactor Rwork: 0.1939 / FOM work R set: 0.8367 / SU B: 5.86 / SU ML: 0.088 / SU R Cruickshank DPI: 0.0386 / SU Rfree: 0.0324 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.039 / ESU R Free: 0.032 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : RESIDUAL ONLY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.7 Å / Shrinkage radii: 0.7 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 72.75 Å2 / Biso mean: 25.435 Å2 / Biso min: 12.37 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2→43.86 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.052 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
