| Entry | Database: PDB / ID: 4nwj
|
|---|
| Title | Crystal structure of phosphopglycerate mutase from Staphylococcus aureus in 3-phosphoglyceric acid bound form. |
|---|
 Components Components | 2,3-bisphosphoglycerate-independent phosphoglycerate mutase |
|---|
 Keywords Keywords | ISOMERASE / glycolytic enzyme / cytosol |
|---|
| Function / homology |  Function and homology information Function and homology information
phosphoglycerate mutase (2,3-diphosphoglycerate-independent) / regulation of sporulation / phosphoglycerate mutase activity / glucose catabolic process / glycolytic process / manganese ion binding / carbohydrate metabolic process / cytosolSimilarity search - Function 2,3-Bisphosphoglycerate-independent phosphoglycerate mutase, substrate-binding domain / BPG-independent phosphoglycerate mutase, domain B / Phosphoglycerate mutase, 2,3-bisphosphoglycerate-independent / BPG-independent PGAM, N-terminal / BPG-independent phosphoglycerate mutase, domain B superfamily / BPG-independent PGAM N-terminus (iPGM_N) / Metalloenzyme / Metalloenzyme superfamily / Alkaline Phosphatase, subunit A / Alkaline Phosphatase, subunit A ...2,3-Bisphosphoglycerate-independent phosphoglycerate mutase, substrate-binding domain / BPG-independent phosphoglycerate mutase, domain B / Phosphoglycerate mutase, 2,3-bisphosphoglycerate-independent / BPG-independent PGAM, N-terminal / BPG-independent phosphoglycerate mutase, domain B superfamily / BPG-independent PGAM N-terminus (iPGM_N) / Metalloenzyme / Metalloenzyme superfamily / Alkaline Phosphatase, subunit A / Alkaline Phosphatase, subunit A / Alkaline-phosphatase-like, core domain superfamily / 3-Layer(aba) Sandwich / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |   Staphylococcus aureus subsp. aureus (bacteria) Staphylococcus aureus subsp. aureus (bacteria) |
|---|
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.01 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.01 Å |
|---|
 Authors Authors | Roychowdhury, A. / Bose, M. / Kundu, A. / Gujar, A. / Das, A.K. |
|---|
 Citation Citation | Journal: Febs J. / Year: 2015
Title: Complete catalytic cycle of cofactor-independent phosphoglycerate mutase involves a spring-loaded mechanism
Authors: Roychowdhury, A. / Kundu, A. / Bose, M. / Gujar, A. / Mukherjee, S. / Das, A.K. |
|---|
| History | | Deposition | Dec 6, 2013 | Deposition site: RCSB / Processing site: PDBJ |
|---|
| Revision 1.0 | Jan 14, 2015 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Feb 11, 2015 | Group: Database references |
|---|
| Revision 1.2 | Apr 8, 2015 | Group: Database references |
|---|
| Revision 1.3 | Nov 8, 2023 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj
 Assembly
Assembly

 Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn
Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn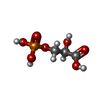
 Mass: 186.057 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7O7P
Mass: 186.057 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7O7P Mass: 18.015 Da / Num. of mol.: 296 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 296 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing