| 登録情報 | データベース: PDB / ID: 4npe
|
|---|
| タイトル | High-resolution structure of C domain of staphylococcal protein A at room temperature |
|---|
 要素 要素 | Immunoglobulin G-binding protein A |
|---|
 キーワード キーワード | PROTEIN BINDING / SpA / three-helix bundle / conformational heterogeneity / rapidly unfolding and folding / RUF / Antibody binding / Antibody |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
Immunoglobulin FC, subunit C / Octapeptide repeat / Octapeptide repeat / Protein A, Ig-binding domain / B domain / Lysin motif / LysM domain superfamily / LysM domain / LysM domain profile. / LysM domain ...Immunoglobulin FC, subunit C / Octapeptide repeat / Octapeptide repeat / Protein A, Ig-binding domain / B domain / Lysin motif / LysM domain superfamily / LysM domain / LysM domain profile. / LysM domain / Immunoglobulin/albumin-binding domain superfamily / YSIRK type signal peptide / YSIRK Gram-positive signal peptide / LPXTG cell wall anchor motif / Gram-positive cocci surface proteins LPxTG motif profile. / LPXTG cell wall anchor domain / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / Up-down Bundle / Mainly Alpha類似検索 - ドメイン・相同性 THIOCYANATE ION / Immunoglobulin G-binding protein A類似検索 - 構成要素 |
|---|
| 生物種 |   Staphylococcus aureus (黄色ブドウ球菌) Staphylococcus aureus (黄色ブドウ球菌) |
|---|
| 手法 |  X線回折 / シンクロトロン / 単波長異常分散 / 解像度: 1.42 Å X線回折 / シンクロトロン / 単波長異常分散 / 解像度: 1.42 Å |
|---|
 データ登録者 データ登録者 | Deis, L.N. / Pemble IV, C.W. / Oas, T.G. / Richardson, J.S. / Richardson, D.C. |
|---|
 引用 引用 | ジャーナル: Structure / 年: 2014
タイトル: Multiscale conformational heterogeneity in staphylococcal protein a: possible determinant of functional plasticity.
著者: Deis, L.N. / Pemble, C.W. / Qi, Y. / Hagarman, A. / Richardson, D.C. / Richardson, J.S. / Oas, T.G. |
|---|
| 履歴 | | 登録 | 2013年11月21日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2014年10月8日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2014年10月22日 | Group: Database references |
|---|
| 改定 1.2 | 2017年11月22日 | Group: Refinement description / カテゴリ: software |
|---|
| 改定 1.3 | 2024年2月28日 | Group: Data collection / Database references / Derived calculations
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_struct_conn_angle / struct_conn / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_alt_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_alt_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_ptnr1_label_alt_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード









 PDBj
PDBj


 集合体
集合体

 分子量: 65.409 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Zn
分子量: 65.409 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Zn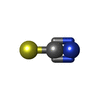
 分子量: 58.082 Da / 分子数: 1 / 由来タイプ: 合成 / 式: CNS
分子量: 58.082 Da / 分子数: 1 / 由来タイプ: 合成 / 式: CNS 分子量: 18.015 Da / 分子数: 50 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 50 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 22-ID / 波長: 1 Å
/ ビームライン: 22-ID / 波長: 1 Å 解析
解析