 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4mb4: Crystal structure of E153Q mutant of cold-adapted chitinase from ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4mb4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of E153Q mutant of cold-adapted chitinase from Moritella complex with Nag4 | |||||||||
 Components Components | Chitinase 60 | |||||||||
 Keywords Keywords | HYDROLASE / TIM-barrel / alpha/beta-barrel Ig-like / Immunoglobulin like domain / ChBD / Chitin binding domain / Nag4 / Chitinase / hydrolaze / low activity mutant | |||||||||
| Function / homology |  Function and homology information Function and homology informationendochitinase activity / chitinase / chitin binding / carbohydrate binding / carbohydrate metabolic process / extracellular region Similarity search - Function | |||||||||
| Biological species |  Moritella marina (bacteria) Moritella marina (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.481 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.481 Å | |||||||||
 Authors Authors | Malecki, P.H. / Vorgias, C.E. / Rypniewski, W. | |||||||||
 Citation Citation | Journal: Acta Crystallogr D Biol Crystallogr / Year: 2014 Title: Crystal structures of substrate-bound chitinase from the psychrophilic bacterium Moritella marina and its structure in solution. Authors: Piotr H Malecki / Constantinos E Vorgias / Maxim V Petoukhov / Dmitri I Svergun / Wojciech Rypniewski /    Abstract: The four-domain structure of chitinase 60 from Moritella marina (MmChi60) is outstanding in its complexity. Many glycoside hydrolases, such as chitinases and cellulases, have multi-domain structures, ...The four-domain structure of chitinase 60 from Moritella marina (MmChi60) is outstanding in its complexity. Many glycoside hydrolases, such as chitinases and cellulases, have multi-domain structures, but only a few have been solved. The flexibility of the hinge regions between the domains apparently makes these proteins difficult to crystallize. The analysis of an active-site mutant of MmChi60 in an unliganded form and in complex with the substrates NAG4 and NAG5 revealed significant differences in the substrate-binding site compared with the previously determined complexes of most studied chitinases. A SAXS experiment demonstrated that in addition to the elongated state found in the crystal, the protein can adapt other conformations in solution ranging from fully extended to compact. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2013Title: Structure of a complete four-domain chitinase from Moritella marina, a marine psychrophilic bacterium Authors: Malecki, P.H. / Raczynska, J.E. / Vorgias, C.E. / Rypniewski, W. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4mb4.cif.gz | 246.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4mb4.ent.gz | 194.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4mb4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4mb4_validation.pdf.gz | 856 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4mb4_full_validation.pdf.gz | 861.4 KB | Display | |
| Data in XML | 4mb4_validation.xml.gz | 28.8 KB | Display | |
| Data in CIF | 4mb4_validation.cif.gz | 45.5 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mb/4mb4ftp://data.pdbj.org/pub/pdb/validation_reports/mb/4mb4 | HTTPS FTP |
-Related structure data
| Related structure data |  4mb3C  4mb5C  4hmcS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein / Sugars , 2 types, 2 molecules A
| #1: Protein | Mass: 58616.371 Da / Num. of mol.: 1 / Mutation: E153Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) Moritella marina (bacteria) / Gene: chi60 / Plasmid: pET-11a / Production host: |
|---|---|
| #2: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2- ...2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source |
-Non-polymers , 5 types, 695 molecules 


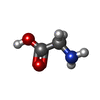

| #3: Chemical |  Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#5: Chemical | ChemComp-SO4 / |  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#6: Chemical |  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H5NO2#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 687 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|---|
| Sequence details | THE SEQUENCE DISCREPANCIES (R452H, A470T) WERE NOT INTENTIONAL MUTATIONS, AND COULD BE THE CORRECT ...THE SEQUENCE DISCREPANC |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.85 Å3/Da / Density % sol: 56.9 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 12.5% w/v PEG 1000, 12.5% w/v PEG 3350, 12.5% v/v MPD, 0.02M Na-L-glutamate, 0.02M alanine (racemic), 0.02M glycine, 0.02M lysine HCl (racemic), 0.02M serine (racemic), 0.1M MES/imidazole pH ...Details: 12.5% w/v PEG 1000, 12.5% w/v PEG 3350, 12.5% v/v MPD, 0.02M Na-L-glutamate, 0.02M alanine (racemic), 0.02M glycine, 0.02M lysine HCl (racemic), 0.02M serine (racemic), 0.1M MES/imidazole pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 292K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY / Beamline: 14.2 / Wavelength: 0.91841 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RAYONIX MX-225 / Detector: CCD / Date: Apr 21, 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Double-crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.91841 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Number: 758690 / Rmerge(I) obs: 0.054 / D res high: 1.48 Å / Num. obs: 110785 / % possible obs: 99.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.48→33.371 Å / Num. all: 110794 / Num. obs: 110794 / % possible obs: 99.8 % / Observed criterion σ(I): -3 / Redundancy: 6.9 % / Biso Wilson estimate: 16.83 Å2 / Rmerge(I) obs: 0.054 / Net I/σ(I): 20.52 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 1.48→1.57 Å / Redundancy: 6.77 % / Rmerge(I) obs: 0.853 / Mean I/σ(I) obs: 2.02 / % possible all: 99.4 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4HMC Resolution: 1.481→33.371 Å / Occupancy max: 1 / Occupancy min: 0 / SU ML: 0.14 / σ(F): 1.99 / Phase error: 18.75 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 65.49 Å2 / Biso mean: 25.3686 Å2 / Biso min: 10.13 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.14 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.481→33.371 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 8
|
