 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4lvz | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the THF riboswitch bound to 2,6-diaminopurine | ||||||
 Components Components | THF riboswitch | ||||||
 Keywords Keywords | RNA / Aptamers / Nucleotide / Bacillus subtilis / Bacterial Proteins / Base Sequence / Binding Sites / Calorimetry / Folic Acid / Gene Expression Regulation / Bacterial / Guanine / Leucovorin / Ligands / Magnesium / Molecular Sequence Data / Nucleic Acid Conformation / Point Mutation / Protein Binding / Protein Structure / Secondary / Riboswitch / S-Adenosylmethionine / Streptococcus mutans / Terminator Regions / Genetic / Tetrahydrofolates / Thermodynamics / Transcription / three-way junction / pseudoknot / regulation / ncRNA / 2 / 6-diaminopurine / mRNA | ||||||
| Function / homology | 9H-PURINE-2,6-DIAMINE / RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.77 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.77 Å | ||||||
 Authors Authors | Trausch, J.J. / Batey, R.T. | ||||||
 Citation Citation | Journal: Chem.Biol. / Year: 2014 Title: A Disconnect between High-Affinity Binding and Efficient Regulation by Antifolates and Purines in the Tetrahydrofolate Riboswitch. Authors: Trausch, J.J. / Batey, R.T. #1: Journal: Structure / Year: 2011Title: The structure of a tetrahydrofolate-sensing riboswitch reveals two ligand binding sites in a single aptamer. Authors: Trausch, J.J. / Ceres, P. / Reyes, F.E. / Batey, R.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4lvz.cif.gz | 66.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4lvz.ent.gz | 48.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4lvz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4lvz_validation.pdf.gz | 402.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4lvz_full_validation.pdf.gz | 403.4 KB | Display | |
| Data in XML | 4lvz_validation.xml.gz | 8.9 KB | Display | |
| Data in CIF | 4lvz_validation.cif.gz | 13.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lv/4lvzftp://data.pdbj.org/pub/pdb/validation_reports/lv/4lvz | HTTPS FTP |
-Related structure data













-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: RNA chain | Mass: 28849.145 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: T7 in vitro transcribed |
|---|---|
| #2: Chemical | ChemComp-6AP / 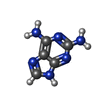  Mass: 150.141 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6N6 Mass: 150.141 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H6N6 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 307 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 307 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.51 Å3/Da / Density % sol: 51.07 % |
|---|---|
| Crystal grow | Temperature: 303 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 2,4-methyl-pentanediol, spermine, sodium cacodylate, sodium chloride, dithiothreitol, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 303K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 1.1051 Å / Beamline: 5.0.2 / Wavelength: 1.1051 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jan 19, 2013 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1051 Å / Relative weight: 1 |
| Reflection | Resolution: 1.77→41.74 Å / Num. all: 28959 / % possible obs: 98.7 % / Observed criterion σ(F): 4.4 / Observed criterion σ(I): 4.4 / Redundancy: 3.3 % |
| Reflection shell | Resolution: 1.77→1.86 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.185 / Mean I/σ(I) obs: 4.4 / Num. unique all: 4204 / Rsym value: 0.17 / % possible all: 99.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.77→28.71 Å / SU ML: 0.23 / σ(F): 1.35 / Phase error: 28.36 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.77→28.71 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
