 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ll5: Crystal Structure of Pim-1 in complex with the fluorescent compou... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ll5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Pim-1 in complex with the fluorescent compound SKF86002 | ||||||
 Components Components | Serine/threonine-protein kinase pim-1 | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / co-crystallization / SKF86002 / fluorescence / inhibitor screening / kinase / TRANSFERASE-TRANSFERASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of cardioblast proliferation / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / ribosomal small subunit binding / positive regulation of cyclin-dependent protein serine/threonine kinase activity / positive regulation of cardiac muscle cell proliferation / negative regulation of innate immune response ...positive regulation of cardioblast proliferation / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / ribosomal small subunit binding / positive regulation of cyclin-dependent protein serine/threonine kinase activity / positive regulation of cardiac muscle cell proliferation / negative regulation of innate immune response / positive regulation of brown fat cell differentiation / Signaling by FLT3 fusion proteins / positive regulation of TORC1 signaling / protein serine/threonine kinase activator activity / regulation of transmembrane transporter activity / positive regulation of protein serine/threonine kinase activity / negative regulation of DNA-binding transcription factor activity / cellular response to type II interferon / manganese ion binding / Interleukin-4 and Interleukin-13 signaling / protein autophosphorylation / non-specific serine/threonine protein kinase / protein stabilization / protein phosphorylation / protein serine kinase activity / protein serine/threonine kinase activity / negative regulation of apoptotic process / nucleolus / apoptotic process / positive regulation of DNA-templated transcription / nucleoplasm / ATP binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2 Å | ||||||
 Authors Authors | Parker, L.J. / Tanaka, A. / Handa, N. / Honda, K. / Tomabechi, Y. / Shirouzu, M. / Yokoyama, S. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2014 Title: Kinase crystal identification and ATP-competitive inhibitor screening using the fluorescent ligand SKF86002. Authors: Parker, L.J. / Taruya, S. / Tsuganezawa, K. / Ogawa, N. / Mikuni, J. / Honda, K. / Tomabechi, Y. / Handa, N. / Shirouzu, M. / Yokoyama, S. / Tanaka, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ll5.cif.gz | 133.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ll5.ent.gz | 101.1 KB | Display | PDB format |
| PDBx/mmJSON format | 4ll5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4ll5_validation.pdf.gz | 773.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4ll5_full_validation.pdf.gz | 773.4 KB | Display | |
| Data in XML | 4ll5_validation.xml.gz | 13.6 KB | Display | |
| Data in CIF | 4ll5_validation.cif.gz | 19.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ll/4ll5ftp://data.pdbj.org/pub/pdb/validation_reports/ll/4ll5 | HTTPS FTP |
-Related structure data
| Related structure data |  4lm5C  4lmuC  4ludC  4lueC  3uixS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34187.727 Da / Num. of mol.: 1 / Fragment: Protein Kinase domain, UNP resodies 120-404 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PIM1 / Plasmid: pCR2.1-TOPO / Production host:  References: UniProt: P11309, non-specific serine/threonine protein kinase | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-SK8 / 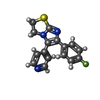  Mass: 297.350 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H12FN3S Mass: 297.350 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H12FN3S | ||||
| #3: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#4: Chemical | ChemComp-CA / |   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 173 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 173 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.3 Å3/Da / Density % sol: 62.7 % / Mosaicity: 0.13 ° |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: 100mM CITRATE BUFFER PH 5.5, 200mM NACL, 1M NH4HPO4, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron  / Beamline: MX2 / Wavelength: 0.95 Å / Beamline: MX2 / Wavelength: 0.95 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Sep 16, 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.95 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→85.433 Å / Num. obs: 29928 / % possible obs: 99.4 % / Redundancy: 11.2 % / Biso Wilson estimate: 26.29 Å2 / Rsym value: 0.115 / Net I/σ(I): 15.6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3UIX Resolution: 2→42.717 Å / Occupancy max: 1 / Occupancy min: 0.4 / FOM work R set: 0.8914 / SU ML: 0.13 / σ(F): 1.36 / Phase error: 18.18 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 95.64 Å2 / Biso mean: 37.1657 Å2 / Biso min: 14.04 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→42.717 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 11
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
