| Entry | Database: PDB / ID: 4ku8
|
|---|
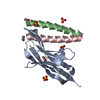
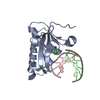
| Title | Structures of PKGI Reveal a cGMP-Selective Activation Mechanism |
|---|
 Components Components | cGMP-dependent Protein Kinase 1 |
|---|
 Keywords Keywords | SIGNALING PROTEIN / cyclic nucleotide binding domain / cGMP |
|---|
| Function / homology |  Function and homology information Function and homology information
cGMP-dependent protein kinase / cGMP-dependent protein kinase activity / collateral sprouting / bone growth / receptor guanylyl cyclase signaling pathway / negative regulation of platelet aggregation / relaxation of vascular associated smooth muscle / Rap1 signalling / regulation of vascular permeability / cGMP effects ...cGMP-dependent protein kinase / cGMP-dependent protein kinase activity / collateral sprouting / bone growth / receptor guanylyl cyclase signaling pathway / negative regulation of platelet aggregation / relaxation of vascular associated smooth muscle / Rap1 signalling / regulation of vascular permeability / cGMP effects / negative regulation of vascular associated smooth muscle cell migration / Maturation of DENV proteins / cGMP binding / negative regulation of vascular associated smooth muscle cell proliferation / calcium channel regulator activity / Ca2+ pathway / protein kinase activity / protein serine kinase activity / Golgi apparatus / signal transduction / ATP binding / identical protein binding / plasma membrane / cytosol / cytoplasmSimilarity search - Function cGMP-dependent protein kinase, N-terminal coiled-coil domain / Coiled-coil N-terminus of cGMP-dependent protein kinase / cGMP-dependent kinase / cGMP-dependent protein kinase, catalytic domain / Cyclic nucleotide-binding domain signature 2. / Cyclic nucleotide-binding domain signature 1. / Cyclic nucleotide-binding, conserved site / Cyclic nucleotide-monophosphate binding domain / Cyclic nucleotide-binding domain / cAMP/cGMP binding motif profile. ...cGMP-dependent protein kinase, N-terminal coiled-coil domain / Coiled-coil N-terminus of cGMP-dependent protein kinase / cGMP-dependent kinase / cGMP-dependent protein kinase, catalytic domain / Cyclic nucleotide-binding domain signature 2. / Cyclic nucleotide-binding domain signature 1. / Cyclic nucleotide-binding, conserved site / Cyclic nucleotide-monophosphate binding domain / Cyclic nucleotide-binding domain / cAMP/cGMP binding motif profile. / Cyclic nucleotide-binding domain / Cyclic nucleotide-binding domain superfamily / Jelly Rolls / Extension to Ser/Thr-type protein kinases / AGC-kinase, C-terminal / AGC-kinase C-terminal domain profile. / RmlC-like jelly roll fold / Jelly Rolls / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / Sandwich / Mainly BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.994 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.994 Å |
|---|
 Authors Authors | Huang, G.Y. / Kim, J.J. / Reger, A.S. / Lorenz, R. / Moon, E.W. / Casteel, D.E. / Sankaran, B. / Herberg, F.W. / Kim, C. |
|---|
 Citation Citation | Journal: Structure / Year: 2014
Title: Structural Basis for Cyclic-Nucleotide Selectivity and cGMP-Selective Activation of PKG I.
Authors: Huang, G.Y. / Kim, J.J. / Reger, A.S. / Lorenz, R. / Moon, E.W. / Zhao, C. / Casteel, D.E. / Bertinetti, D. / Vanschouwen, B. / Selvaratnam, R. / Pflugrath, J.W. / Sankaran, B. / Melacini, G. ...Authors: Huang, G.Y. / Kim, J.J. / Reger, A.S. / Lorenz, R. / Moon, E.W. / Zhao, C. / Casteel, D.E. / Bertinetti, D. / Vanschouwen, B. / Selvaratnam, R. / Pflugrath, J.W. / Sankaran, B. / Melacini, G. / Herberg, F.W. / Kim, C. |
|---|
| History | | Deposition | May 21, 2013 | Deposition site: RCSB / Processing site: RCSB |
|---|
| Revision 1.0 | Jan 15, 2014 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jan 29, 2014 | Group: Database references |
|---|
| Revision 1.2 | Nov 15, 2017 | Group: Refinement description / Category: software |
|---|
| Revision 1.3 | Feb 28, 2024 | Group: Data collection / Database references / Derived calculations
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj








 Assembly
Assembly




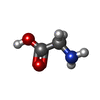
 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H5NO2
Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H5NO2 Mass: 18.015 Da / Num. of mol.: 344 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 344 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 31-ID / Wavelength: 0.97931 Å
/ Beamline: 31-ID / Wavelength: 0.97931 Å Processing
Processing