- PDB-4io8: Crystal structure of human HSP70 complexed with 4-{(2R,3S,4R)-5-[... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4io8
Title
Crystal structure of human HSP70 complexed with 4-{(2R,3S,4R)-5-[(R)-6-Amino-8-(3,4-dichloro-benzylamino)-purin-9-yl]-3,4-dihydroxy-tetrahydro-furan-2-ylmethoxymethyl}-benzonitrile
Components
Heat shock 70kDa protein 1A variant
Keywords
CHAPERONE / Hsp70 / ATPase / molecular chaperone
Function / homology
Function and homology information
: / negative regulation of inclusion body assembly / cellular heat acclimation / positive regulation of nucleotide-binding oligomerization domain containing 2 signaling pathway / Viral RNP Complexes in the Host Cell Nucleus / C3HC4-type RING finger domain binding / positive regulation of microtubule nucleation / ATP-dependent protein disaggregase activity / misfolded protein binding / negative regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway ...: / negative regulation of inclusion body assembly / cellular heat acclimation / positive regulation of nucleotide-binding oligomerization domain containing 2 signaling pathway / Viral RNP Complexes in the Host Cell Nucleus / C3HC4-type RING finger domain binding / positive regulation of microtubule nucleation / ATP-dependent protein disaggregase activity / misfolded protein binding / negative regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway / aggresome / regulation of mitotic spindle assembly / positive regulation of tumor necrosis factor-mediated signaling pathway / lysosomal transport / cellular response to steroid hormone stimulus / mRNA catabolic process / Dengue Virus Genome Translation and Replication / cellular response to unfolded protein / regulation of protein ubiquitination / HSF1-dependent transactivation / negative regulation of extrinsic apoptotic signaling pathway in absence of ligand / Regulation of HSF1-mediated heat shock response / response to unfolded protein / Mitochondrial unfolded protein response (UPRmt) / Attenuation phase / chaperone-mediated protein complex assembly / negative regulation of endoplasmic reticulum stress-induced intrinsic apoptotic signaling pathway / transcription regulator inhibitor activity / ATP metabolic process / endoplasmic reticulum unfolded protein response / heat shock protein binding / positive regulation of erythrocyte differentiation / negative regulation of protein ubiquitination / centriole / protein folding chaperone / inclusion body / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / positive regulation of RNA splicing / positive regulation of interleukin-8 production / negative regulation of transforming growth factor beta receptor signaling pathway / AUF1 (hnRNP D0) binds and destabilizes mRNA / negative regulation of cell growth / PKR-mediated signaling / G protein-coupled receptor binding / histone deacetylase binding / disordered domain specific binding / : / transcription corepressor activity / positive regulation of proteasomal ubiquitin-dependent protein catabolic process / cellular response to heat / protein refolding / virus receptor activity / cellular response to oxidative stress / blood microparticle / vesicle / ficolin-1-rich granule lumen / positive regulation of canonical NF-kappaB signal transduction / nuclear speck / protein stabilization / cadherin binding / negative regulation of cell population proliferation / receptor ligand activity / ribonucleoprotein complex / signaling receptor binding / focal adhesion / centrosome / positive regulation of gene expression / ubiquitin protein ligase binding / Neutrophil degranulation / negative regulation of apoptotic process / perinuclear region of cytoplasm / negative regulation of transcription by RNA polymerase II / enzyme binding / endoplasmic reticulum / ATP hydrolysis activity / protein-containing complex / mitochondrion / : / RNA binding / extracellular exosome / extracellular region / nucleoplasm / ATP binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function
Defensin A-like - #30 / Defensin A-like / Heat shock hsp70 proteins family signature 2. / Heat shock hsp70 proteins family signature 1. / Heat shock hsp70 proteins family signature 3. / Heat shock protein 70, conserved site / Heat shock protein 70kD, peptide-binding domain superfamily / Heat shock protein 70kD, C-terminal domain superfamily / Heat shock protein 70 family / Hsp70 protein ...Defensin A-like - #30 / Defensin A-like / Heat shock hsp70 proteins family signature 2. / Heat shock hsp70 proteins family signature 1. / Heat shock hsp70 proteins family signature 3. / Heat shock protein 70, conserved site / Heat shock protein 70kD, peptide-binding domain superfamily / Heat shock protein 70kD, C-terminal domain superfamily / Heat shock protein 70 family / Hsp70 protein / ATPase, substrate binding domain, subdomain 4 / Actin; Chain A, domain 4 / ATPase, nucleotide binding domain / ATPase, nucleotide binding domain / Nucleotidyltransferase; domain 5 / Alpha-Beta Complex / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj








 Assembly
Assembly

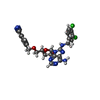
 Mass: 556.401 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H23Cl2N7O4
Mass: 556.401 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H23Cl2N7O4 Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X06SA / Wavelength: 1 Å
/ Beamline: X06SA / Wavelength: 1 Å Processing
Processing