 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ine: Crystal structure of N-methyl transferase (PMT-2) from Caenorhabd... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ine | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of N-methyl transferase (PMT-2) from Caenorhabditis elegant complexed with S-adenosyl homocysteine and phosphoethanolamine | ||||||
 Components Components | Protein PMT-2 | ||||||
 Keywords Keywords | TRANSFERASE / methyl transferase | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphatidyl-N-methylethanolamine N-methyltransferase activity / phosphoethanolamine N-methyltransferase activity / phosphoethanolamine N-methyltransferase / N-methyltransferase activity / phosphatidylcholine biosynthetic process / methylation Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | ||||||
 Authors Authors | Lukk, T. / Nair, S.K. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure of N-methyl transferase (PMT-2) from Caenorhabditis elegant complexed with S-adenosyl homocysteine and phosphoethanolamine Authors: Lukk, T. / Nair, S.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ine.cif.gz | 212.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ine.ent.gz | 165.8 KB | Display | PDB format |
| PDBx/mmJSON format | 4ine.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/in/4ineftp://data.pdbj.org/pub/pdb/validation_reports/in/4ine | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3ujbS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj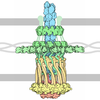



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 51710.957 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical |   Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20N6O5S Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H20N6O5S#3: Chemical | 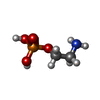  Mass: 141.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H8NO4P Mass: 141.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H8NO4P#4: Chemical |   Mass: 78.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6OS Mass: 78.133 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6OS#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1086 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1086 / Source method: isolated from a natural source / Formula: H2OSequence details | THE SPECIFIC CLONE IS AN ISOFORM OF THE SEQUENCE OF UNP ENTRY Q22993. RESIDUES LYS 89 AND ASN 93 ...THE SPECIFIC CLONE IS AN ISOFORM OF THE SEQUENCE OF UNP ENTRY Q22993. RESIDUES LYS 89 AND ASN 93 ARE LINKED TOGETHER | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.8 % |
|---|---|
| Crystal grow | Temperature: 282 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: Protein solution was at 15 mg/mL containing 20 mM Tris (pH 7.5), 100 mM NaCl, 10 mM EDTA and 10 mM bME, 5 mM SAH, 5 mM phosphoethanolamine. Mother liqueur contained 0.04 M potassium ...Details: Protein solution was at 15 mg/mL containing 20 mM Tris (pH 7.5), 100 mM NaCl, 10 mM EDTA and 10 mM bME, 5 mM SAH, 5 mM phosphoethanolamine. Mother liqueur contained 0.04 M potassium phosphate (monobasic), 16% PEG 8,000 and 20% v/v glycerol, VAPOR DIFFUSION, SITTING DROP, temperature 282K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-G / Wavelength: 0.97857 / Beamline: 21-ID-G / Wavelength: 0.97857 |
| Detector | Type: RAYONIX MX-300 / Detector: CCD / Date: Jun 3, 2012 |
| Radiation | Monochromator: C(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→30 Å / Num. obs: 172409 / % possible obs: 96.5 % / Observed criterion σ(I): 0 / Redundancy: 4.3 % / Rsym value: 0.05 / Net I/σ(I): 11.6 |
| Reflection shell | Resolution: 1.45→1.5 Å / Redundancy: 3.6 % / Rmerge(I) obs: 0.634 / % possible all: 93.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 3UJB Resolution: 1.45→29.63 Å / Cor.coef. Fo:Fc: 0.968 / Cor.coef. Fo:Fc free: 0.96 / Occupancy max: 1 / Occupancy min: 0.38 / SU B: 1.059 / SU ML: 0.041 / SU R Cruickshank DPI: 0.0713 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.071 / ESU R Free: 0.071 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.18 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→29.63 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.45→1.49 Å / Total num. of bins used: 20
|
