 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4igk | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human BRCA1 BRCT in complex with ATRIP peptide | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION / Cell cycle / Disease mutation / DNA damage / DNA repair / Fatty acid biosynthesis / Ligase / Lipid synthesis / Metal-binding / Nucleus / Phosphoprotein / Tumor suppressor / Ubl conjugation pathway / Zinc-finger / BRCT domain / DNA damage response / phospho peptide interactions / DNA-binding / phospho peptide binding | ||||||
| Function / homology |  Function and homology information Function and homology informationATR-ATRIP complex / histone H2AK127 ubiquitin ligase activity / histone H2AK129 ubiquitin ligase activity / Defective DNA double strand break response due to BRCA1 loss of function / Defective DNA double strand break response due to BARD1 loss of function / BRCA1-BARD1 complex / BRCA1-B complex / BRCA1-C complex / BRCA1-A complex / negative regulation of centriole replication ...ATR-ATRIP complex / histone H2AK127 ubiquitin ligase activity / histone H2AK129 ubiquitin ligase activity / Defective DNA double strand break response due to BRCA1 loss of function / Defective DNA double strand break response due to BARD1 loss of function / BRCA1-BARD1 complex / BRCA1-B complex / BRCA1-C complex / BRCA1-A complex / negative regulation of centriole replication / random inactivation of X chromosome / gamma-tubulin ring complex / negative regulation of intracellular estrogen receptor signaling pathway / ubiquitin-modified histone reader activity / chordate embryonic development / cellular response to indole-3-methanol / DNA strand resection involved in replication fork processing / nuclear ubiquitin ligase complex / homologous recombination / negative regulation of fatty acid biosynthetic process / Regulation of MITF-M-dependent genes involved in DNA replication, damage repair and senescence / lateral element / mitotic G2/M transition checkpoint / XY body / Impaired BRCA2 binding to PALB2 / regulation of DNA damage checkpoint / regulation of double-strand break repair / DNA repair complex / RNA polymerase binding / DNA damage tolerance / protein K6-linked ubiquitination / nucleobase-containing compound metabolic process / centrosome cycle / K63-linked polyubiquitin modification-dependent protein binding / HDR through Single Strand Annealing (SSA) / response to ionizing radiation / Homologous DNA Pairing and Strand Exchange / Defective homologous recombination repair (HRR) due to BRCA1 loss of function / Defective HDR through Homologous Recombination Repair (HRR) due to PALB2 loss of BRCA1 binding function / Defective HDR through Homologous Recombination Repair (HRR) due to PALB2 loss of BRCA2/RAD51/RAD51C binding function / Resolution of D-loop Structures through Synthesis-Dependent Strand Annealing (SDSA) / negative regulation of gene expression via chromosomal CpG island methylation / Resolution of D-loop Structures through Holliday Junction Intermediates / mitotic G2 DNA damage checkpoint signaling / Transcriptional Regulation by E2F6 / Impaired BRCA2 binding to RAD51 / negative regulation of cell cycle / negative regulation of reactive oxygen species metabolic process / positive regulation of vascular endothelial growth factor production / Presynaptic phase of homologous DNA pairing and strand exchange / Activation of ATR in response to replication stress / regulation of DNA repair / negative regulation of extrinsic apoptotic signaling pathway via death domain receptors / ubiquitin ligase complex / SUMOylation of DNA damage response and repair proteins / protein autoubiquitination / DNA damage checkpoint signaling / Meiotic synapsis / positive regulation of DNA repair / male germ cell nucleus / cellular response to ionizing radiation / TP53 Regulates Transcription of DNA Repair Genes / chromosome segregation / Nonhomologous End-Joining (NHEJ) / Fanconi Anemia Pathway / tubulin binding / negative regulation of cell growth / RING-type E3 ubiquitin transferase / G2/M DNA damage checkpoint / double-strand break repair via homologous recombination / Meiotic recombination / cellular response to tumor necrosis factor / HDR through Homologous Recombination (HRR) / intrinsic apoptotic signaling pathway in response to DNA damage / fatty acid biosynthetic process / Metalloprotease DUBs / positive regulation of angiogenesis / p53 binding / ubiquitin-protein transferase activity / KEAP1-NFE2L2 pathway / double-strand break repair / chromosome / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / Neddylation / Processing of DNA double-strand break ends / Regulation of TP53 Activity through Phosphorylation / damaged DNA binding / transcription coactivator activity / regulation of cell cycle / transcription cis-regulatory region binding / nuclear body / protein ubiquitination / chromatin remodeling / ribonucleoprotein complex / DNA repair / negative regulation of DNA-templated transcription / DNA damage response / ubiquitin protein ligase binding / positive regulation of gene expression / regulation of transcription by RNA polymerase II Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Liu, X. / Ladias, J.A.A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2013 Title: Structural Basis for the BRCA1 BRCT Interaction with the Proteins ATRIP and BAAT1. Authors: Liu, X. / Ladias, J.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4igk.cif.gz | 151.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4igk.ent.gz | 118.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4igk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ig/4igkftp://data.pdbj.org/pub/pdb/validation_reports/ig/4igk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4ifiC  1y98S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

















- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 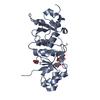
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 24531.234 Da / Num. of mol.: 2 / Fragment: BRCT domain, UNP residues 1646-1859 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BRCA1, RNF53 / Plasmid: pGEX / Production host:  References: UniProt: P38398, Ligases; Forming carbon-nitrogen bonds; Acid-amino-acid ligases (peptide synthases) #2: Protein/peptide | Mass: 788.762 Da / Num. of mol.: 2 / Fragment: UNP residues 237-243 / Source method: obtained synthetically / Details: This sequence occurs naturally in humans. / Source: (synth.) Homo sapiens (human) / References: UniProt: Q8WXE1#3: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 429 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 429 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.27 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 20% PEG3350, 0.2 M ammonium acetate, 8% glycerol, 0.1 M HEPES pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-E / Wavelength: 0.9792 Å / Beamline: 24-ID-E / Wavelength: 0.9792 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Nov 9, 2011 / Details: mirrors |
| Radiation | Monochromator: Fast monochromatic rotary beam shutters / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→50 Å / Num. obs: 43950 / % possible obs: 99 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.6 % / Biso Wilson estimate: 24.4 Å2 / Rsym value: 0.059 / Net I/σ(I): 18.22 |
| Reflection shell | Resolution: 1.75→1.81 Å / Redundancy: 3.6 % / Mean I/σ(I) obs: 7.5 / Num. unique all: 4350 / Rsym value: 0.178 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1Y98 Resolution: 1.75→27.51 Å / Cor.coef. Fo:Fc: 0.973 / Cor.coef. Fo:Fc free: 0.959 / SU B: 2.631 / SU ML: 0.07 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 1.5 / σ(I): 7.5 / ESU R: 0.11 / ESU R Free: 0.112 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.039 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.225 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→27.51 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.751→1.796 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
