 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4hmr | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of mutant rabbit PRP 121-230 (S170N/S174N) | ||||||
 Components Components | Major prion protein | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / prp / prion / Membrane | ||||||
| Function / homology |  Function and homology information Function and homology informationaspartic-type endopeptidase inhibitor activity / regulation of calcium ion import across plasma membrane / glycosaminoglycan binding / regulation of potassium ion transmembrane transport / positive regulation of glutamate receptor signaling pathway / cupric ion binding / negative regulation of interleukin-17 production / type 5 metabotropic glutamate receptor binding / negative regulation of dendritic spine maintenance / negative regulation of calcineurin-NFAT signaling cascade ...aspartic-type endopeptidase inhibitor activity / regulation of calcium ion import across plasma membrane / glycosaminoglycan binding / regulation of potassium ion transmembrane transport / positive regulation of glutamate receptor signaling pathway / cupric ion binding / negative regulation of interleukin-17 production / type 5 metabotropic glutamate receptor binding / negative regulation of dendritic spine maintenance / negative regulation of calcineurin-NFAT signaling cascade / negative regulation of interleukin-2 production / negative regulation of activated T cell proliferation / negative regulation of amyloid-beta formation / negative regulation of type II interferon production / cuprous ion binding / positive regulation of protein targeting to membrane / negative regulation of T cell receptor signaling pathway / side of membrane / positive regulation of calcium-mediated signaling / neuron projection maintenance / inclusion body / cellular response to copper ion / molecular function activator activity / positive regulation of protein localization to plasma membrane / molecular condensate scaffold activity / cellular response to xenobiotic stimulus / protein homooligomerization / protein destabilization / cellular response to amyloid-beta / terminal bouton / positive regulation of neuron apoptotic process / amyloid-beta binding / signaling receptor activity / protease binding / response to oxidative stress / nuclear membrane / microtubule binding / learning or memory / intracellular signal transduction / membrane raft / copper ion binding / dendrite / negative regulation of apoptotic process / protein-containing complex binding / Golgi apparatus / negative regulation of transcription by RNA polymerase II / cell surface / endoplasmic reticulum / identical protein binding / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Sweeting, B. / Chakrabartty, A. / Pai, E.F. | ||||||
 Citation Citation | Journal: Plos One / Year: 2013 Title: N-Terminal Helix-Cap in alpha-Helix 2 Modulates beta-State Misfolding in Rabbit and Hamster Prion Proteins. Authors: Sweeting, B. / Brown, E. / Khan, M.Q. / Chakrabartty, A. / Pai, E.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4hmr.cif.gz | 107.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4hmr.ent.gz | 82.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4hmr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hm/4hmrftp://data.pdbj.org/pub/pdb/validation_reports/hm/4hmr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4hlsC  4hmmC  3o79S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 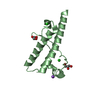
| ||||||||||||
| 3 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 15293.979 Da / Num. of mol.: 2 / Fragment: UNP residues 119-229 / Mutation: S170N, S174N Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical |   Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#3: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#4: Chemical |   Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 124 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 124 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.82 Å3/Da / Density % sol: 32.3 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 100mM SODIUM CACODYLATE PH 6.5, 3.0M NACL, 30% GLYCEROL (cryoprotectant), VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI  / Beamline: 08ID-1 / Wavelength: 0.9793 Å / Beamline: 08ID-1 / Wavelength: 0.9793 Å |
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jul 8, 2008 |
| Radiation | Monochromator: DCM WITH CRYO-COOLED 1ST CRYSTAL, SAGITTALLY BENT 2ND CRYSTAL, FOLLOWED BY VERTICALLY FOCUSING MIRROR Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9793 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→60 Å / Num. all: 30243 / Num. obs: 30243 / % possible obs: 99.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 7.16 % / Rmerge(I) obs: 0.079 / Rsym value: 0.063 / Net I/σ(I): 23.01 |
| Reflection shell | Resolution: 1.6→1.7 Å / Redundancy: 7.3 % / Rmerge(I) obs: 0.372 / Mean I/σ(I) obs: 5.34 / Num. unique all: 4938 / Rsym value: 0.529 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3O79 Resolution: 1.6→61.35 Å / Cor.coef. Fo:Fc: 0.968 / Cor.coef. Fo:Fc free: 0.954 / SU B: 3.073 / SU ML: 0.05 / Isotropic thermal model: Anisotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.102 / ESU R Free: 0.087 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.807 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→61.35 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.642 Å / Total num. of bins used: 20
|
